Complications Associated with Mechanical Ventilation: Introduction
Critically ill patients are at risk of succumbing to their primary disease or the undesired sequelae associated with their therapy. Although frequently lifesaving, the use of positive airway pressure therapy has numerous undesired physiologic and clinical complications. These complications have their origins in the endotracheal or tracheostomy tube, the positive-pressure ventilation (PPV), or from therapies delivered during the care of mechanical ventilation. Other complications may result from coexisting illness or comorbid conditions. Although not commonly recognized as important effects of PPV or positive end-expiratory pressure (PEEP), alterations in organ functions ought to be recognized and addressed accordingly to reduce morbidity and mortality.
Gastrointestinal Tract Complications
The interaction between PPV and the gastrointestinal tract in critical setting is a complex one. Gastrointestinal changes are reported frequently in critically ill patients receiving PPV. The true incidence of gastrointestinal complications is not known, but it is reported to be up to 100% for those receiving PPV for more than 3 days. Splanchnic hypoperfusion seems to play a pivotal role in the pathogenesis of these complications, including mucosal damage, motility disorders, and mesenteric ischemia (Fig. 41-1).1 Unlike many other vascular beds, the splanchnic exhibits limited autoregulation when faced with reduction in mean arterial pressure.2 Without altering total blood flow to the organs of the digestive tract, sympathetic stimulation redistributes blood flow to the muscularis of the wall by decreasing mucosal perfusion.3 This poses severe ischemic changes to the mucosal layer because the mucosa is metabolically more active than the muscle layer and is more vulnerable to the destructive effects of a compromised blood supply.
Figure 41-1
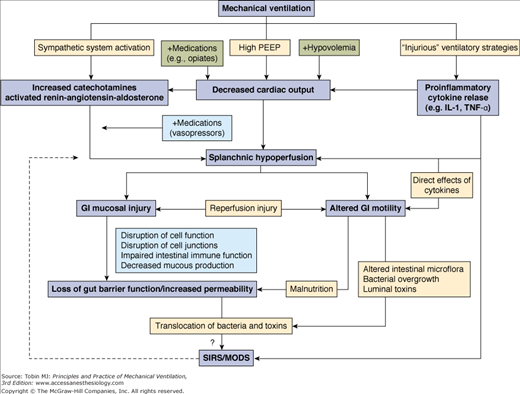
Suggested mechanisms for the development of gastrointestinal (GI) complications during mechanical ventilation. IL, interleukin; MODS, multiple-organ dysfunction syndrome; PEEP, positive end-expiratory pressure; SIRS, systemic inflammatory response syndrome; TNF, tumor necrosis factor. (Used, with permission, from Mutlu.1)
Mechanical ventilation influences the gastrointestinal function by impacting systemic hemodynamics via high PEEP or potentially injurious ventilator strategies such as a high tidal volume (VT). The effect of PEEP on splanchnic blood flow has been shown in animal models to be dose-dependent.4 PEEP decreases venous return and reduces preload, which in turn reduces cardiac output and results in splanchnic hypoperfusion.5 The reduction of splanchnic blood flow is limited at PEEP levels below 10 cm H2O but it is more pronounced at PEEP levels of 15 to 20 cm H2O.6 In rats, the addition of 10 cm H2O of PEEP resulted in reduction of cardiac output and mesenteric blood flow by 31% and 75%, respectively.7 PEEP also promotes plasma-renin-angiotensin-aldosterone activity, as well as catecholamine release, which limits splanchnic hypoperfusion.4,7 Interestingly, high PEEP levels interfere with mesenteric leukocyte–endothelial interaction. In rats with healthy lungs, 10 mbar of PEEP was associated with an increase in the number of rolling, adherent, and migrated leukocytes when compared with anesthesia alone or with mechanical ventilation with 0 or 5 mbar PEEP.8
Alternatively, mechanical ventilation with high VT can modify the inflammatory responses irrespective of the underlying lung injury. Experimental data suggest that mechanical ventilation with high VT and zero end-expiratory pressure induces not only cytokine release (tumor necrosis factor [TNF]-α and interleukin [IL]-8) but also translocation of cytokines from the lungs to the systemic circulation and vice versa.9,10 These cytokines lead to a number of clinical sequelae in the gastrointestinal tract including splanchnic hypoperfusion and intestinal smooth muscle impairment.11,12
Indirectly, medications administered to patients on mechanical ventilation can have deleterious effects on gastrointestinal function. Opiates and sedatives, such as benzodiazepines, can decrease gut motility and impair venous return.13 Other agents, like vasopressors or inotropes,14,15 may alter hemodynamic parameters that, in turn, reduce mesenteric blood flow and put a critically ill patient at risk of developing stress-related mucosal disease.4
Stress-related mucosal disease is the most common cause of gastrointestinal bleeding in patients receiving mechanical ventilation. An estimated 74% to 100% of critically ill patients have endoscopically detectable lesions in the gastric mucosa within hours after admission.16,17 These lesions are most frequently reported in the acid-producing areas of the stomach in contrast to peptic ulcers, which are more common in the antrum and the duodenal bulbs. Overt stress-related gastrointestinal bleeding occurs in up to 5% of critically ill patients.18 Other studies have demonstrated an even lower incidence of clinically significant bleeding, ranging from 0.17% to 1.5%.19 The risk increases with increasing number of days of mechanical ventilation and length of intensive care unit (ICU) stay.20 Other contributing factors include major surgery, head trauma, severe burns, sepsis, glucocorticoids, and renal and hepatic disease on admission.20 Unsurprisingly, clinically significant gastrointestinal bleeding has been linked to prolonged ICU length of stay by as much as 11 days and to a markedly increased mortality, although the primary cause of mortality is attributable to the primary disease process rather than gastrointestinal hemorrhage.20
The pathophysiology of stress-related mucosal disease underlines a complex interaction of opposing vectors (Fig. 41-2).21 Under normal physiologic conditions, the integrity of the mucosa depends on a delicate balance between injurious factors (gastric acid, enzyme secretion) and protective mechanisms (mucous production, prostaglandins).22 Gastric acid is considered essential for stress ulceration but it is not the sole factor in the pathogenesis of mucosal disease. In the setting of decreased splanchnic blood flow, oxygen radicals are released, prostaglandins synthesis is reduced, and nitric oxide production is exaggerated. These changes perpetuate the release of inflammatory cytokines and triggers cell death.23 As a result, increased back diffusion of hydrogen ions and pepsin occurs without the mitigating effect of bicarbonate-rich mucous layer and the protective effect of prostaglandins. Collectively, the imbalance between the noxious gastric acid and the impaired reparative mechanisms predisposes patients on mechanical ventilation to stress-related mucosal injury.
Figure 41-2
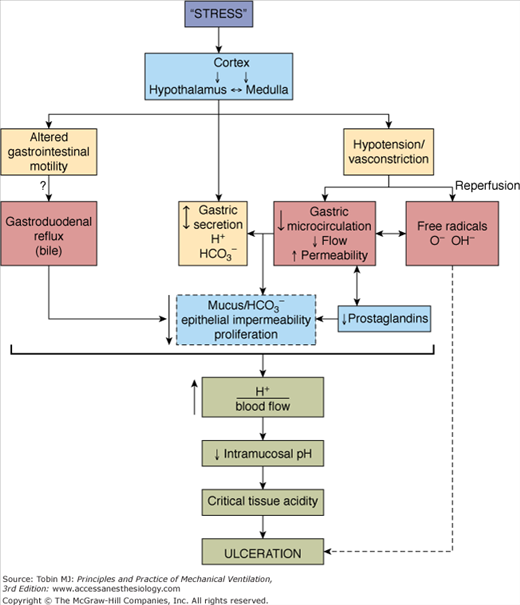
Proposed mechanisms for the development of stress ulceration during mechanical ventilation. (Used, with permission, from Bresalier.21)
The mainstay of the clinical management of stress-related mucosal bleeding in patients who are mechanically ventilated is prevention. Several classes of antistress agents have been introduced since antacids were first evaluated in 1976.24 Antacids directly neutralize luminal gastric acid, bind pepsin and bile acids,25 and may stimulate prostaglandin release from the mucosa.26 The efficacy of antacids in preventing clinically significant stress-related bleeding has been demonstrated in randomized controlled trials.27 The frequent dosing interval needed to achieve and maintain a pH greater than 4, however, is one of the disadvantages of this therapy. In addition to impairing the absorption of other medications, aluminum-containing antacids may be associated with hypophosphatemia and toxic aluminum levels in renal-failure patients, while magnesium-containing antacids may be associated with diarrhea and hypermagnesemia. Sucralfate is another agent that shields the gastric mucosa by forming a protective barrier from the acid in the gastric lumen, stimulating mucous and bicarbonate secretion, and enhancing prostaglandin release.28 A meta-analysis of the efficacy of sucralfate compared with that of histamine-2-receptor antagonists and antacids for the prophylaxis of stress ulcers indicated that sucralfate was at least as effective as the other agents.29 Sucralfate is generally well tolerated but may cause constipation, and aluminium toxicity may occur in patients with renal failure. Misoprostol, a synthetic prostaglandin E1 analog with gastric cytoprotective and antisecretory properties, has also demonstrated effectiveness in preventing gastric injury and complications induced by nonsteroidal antiinflammatory drugs.30 Its use is limited, however, by the high rate of diarrhea and the need for multiple daily doses.
The introduction of histamine-2-receptor antagonists and proton pump inhibitors eclipsed the use of antacids, and medications in these classes are currently the standard therapy for stress-related mucosal disease. Although continuous infusions may be more effective in suppressing gastric acid,31 no studies have demonstrated improved safety, more effective prophylaxis, or a lower rebleeding rate with either method. A major concern of using histamine antagonists is the development of tolerance, which occurs within 42 hours after intravenous administration by both bolus and continuous infusion.32 Histamine antagonists are also associated with adverse chronotropic and inotropic effects and, experimentally, may induce a dose-dependent coronary vasoconstriction.25 The clinical significance of these effects is unclear.
Proton pump inhibitors irreversibly bind the hydrogen-potassium-adenosine triphosphatase, the enzyme responsible for secreting acid into the gastric lumen. Unlike histamine antagonists, proton pump inhibitors do not seem to develop tolerance with sustained therapy. Although both histamine antagonists and proton pump inhibitors can elevate intragastric pH to more than 4, proton pump inhibitors are more likely to maintain a pH above 6.18 Yet in a meta-analysis of randomized, controlled trials that directly compared proton pump inhibitors with histamine antagonists in prevention of stress-related upper gastrointestinal bleeding in ICU patients, both agents were equally efficacious in preventing stress-related bleeding.33
The safety consideration of acid-suppressive therapies has been a concern after several studies suggested that acid-suppressive agents may increase the risk of pneumonia.34,35 Numerous small studies36,37 have examined this issue in ICU patients, but multicenter randomized studies did not demonstrate any significant difference in pneumonia rates between sucralfate and histamine antagonists38 or between proton pump inhibitors and histamine antagonists.33 Other potential adverse effects for acid-suppressing agents include higher rates of Clostridium difficile-associated diarrhea39 and enteric infections.40 None of these associations, however, have been confirmed in randomized trials.
Because acid injury may potentiate mucosal ischemic changes, enteral nutrition could potentially decrease stress ulceration by raising intragastric pH. Several studies in mechanically ventilated patients41 and in burn patients42 have associated enteral feeding with a lower incidence of stress-related bleeding. Other studies in critically ill patients, however, have demonstrated that enteral feeding does not have a significant effect on increasing gastric pH and therefore may be ineffective in affording gastroprotection.43,44 Definite recommendations regarding the role of enteral nutrition for stress-ulcer prophylaxis are not possible at the present because of lack of prospective, randomized trials.
Several studies indicate that abnormal gastrointestinal motility is common in mechanically ventilated patients.45,46 The prevalence of abnormalities in gastric emptying is estimated to be as high as 50%.15 Using manometric evaluation, it was demonstrated that the contractile activity of the stomach is severely depressed in patients receiving PPV—but persistent, albeit reduced, in the duodenum.47 These abnormalities are thought to result from the dysfunction of the interstitial cells of Cajal that act as the controller of gastrointestinal motor activity.48 Several factors are implicated in the pathophysiology of altered gut motility, including preexisting diseases, release of endotoxin49 and corticotropin-releasing factor50 during severe stress, and drugs routinely used in the management of patients on mechanical ventilation (e.g., sedatives and opioid analgesics, catecholamine vasopressors, anticholinergics, and α2-adrenergic receptor agonists). Hyperglycemia has been considered as a risk factor to impair pyloric and antral contractions in healthy volunteers.51 A relationship, however, between hyperglycemia and delayed gastric emptying remains unclear.
Current recommendations for the treatment of impaired gastrointestinal motility focus on optimizing fluid intake, correcting electrolyte disturbances, and minimizing the use of drugs that slow gut motility. Prokinetic agents are not routinely recommended though a recent survey revealed standard prokinetic use in 39% of critically ill patients.52 A combination therapy of erythromycin and metoclopramide provides the most effective regimen in improving the delivery of nasogastric nutrition.53 Owing to a rapid tachyphylaxis following erythromycin or metoclopramide administration, however, therapeutic use should be limited to 3 days. Newer therapeutic approaches, such as μ-opioid receptor antagonists and cholecystokinin-1 receptor antagonists, hold promise in alleviating motility disorders but have not been studied so far in critical illness.
If treatment of gastric stasis fails or is contraindicated, the stomach can be bypassed with an intestinal feeding tube. Early enteral feeding can be more successful if feeds are delivered directly to the small intestine. Postpyloric tubes have been shown to be equally effective as prokinetic treatment in patients who failed nasogastric feeding,54 but there is currently no evidence to support routine use of these tubes in critically ill patients.
Acalculous cholecystitis is a serious and potentially life-threatening illness in critically ill ventilated patients if unrecognized. The condition accounts for 5% to 10% of all cases of acute cholecystitis.55,56 The etiology of the disease remains unknown, although PPV for more than 72 hours is considered a risk factor.55 Other predisposing conditions include shock, dehydration, multiple transfusions, total parenteral nutrition, and drugs (opiates, sedatives, ceftriaxone) (Table 41-1).57,58 Clinical assessment may not be reliable, particularly in intubated and sedated patients. Ultrasonography remains the diagnostic test of choice, although computed tomography has the advantage of being more sensitive in diagnosing acalculous cholecystitis.59,60 Evidence of gallbladder wall thickness greater than or equal to 4 mm, pericholecystic fluid or subserosal edema without ascites, intramural gas, or a sloughed mucosal membrane are considered diagnostic criteria for acute acalculous cholecystitis. Hepatobiliary scintigraphy is compromised by frequent false positives and is more helpful in excluding rather than confirming the diagnosis.59 The mainstay of therapy for acalculous cholecystitis has been cholecystectomy, but for the critically ill patients percutaneous cholecystostomy is considered an alternative to open procedures.61
|
Hepatic Complications
The normal adult liver has a dual blood flow and oxygen supply. Approximately two-thirds of hepatic blood flow and one-half of the oxygen supply is derived from the portal vein while the rest is provided by the hepatic artery. Institution of PPV has significant implications on hepatic perfusion. The reduction in cardiac output observed during PPV causes a proportional drop in global hepatic blood flow.62,63 In addition, the descent of the diaphragm during PPV results in a direct compression of the liver parenchyma and a dramatic rise in intraabdominal pressure. The combination of these two forces leads to an increase in hepatic vascular resistance, which, in turn, impedes portal venous flow. Maintenance of spontaneous breathing during airway pressure release ventilation results in higher hepatic venous oxygen saturation and better hepatic lactate elimination as compared with full ventilator support at equal airway pressure limits.64
The addition of PEEP further complicates the picture. In clinical studies of patients with acute lung injury secondary to septic shock, an increase in PEEP to 15 cm H2O resulted in decrease hepatic vein oxygen saturation compared to 10 cm H2O.65 Similarly, PEEP levels of 15 cm H2O but not 10 cm H2O was associated with a decrease in hepatic glucose production. After liver transplantation, patients are especially vulnerable to change in blood flow because of vascular anastomoses and liver function recovery after cold ischemia. Despite an increase in central venous and pulmonary capillary occluding pressure following 10 mbar of PEEP, there was fortunately no deterioration in Doppler flow velocities of portal and hepatic veins.66
Permissive hypercapnia has been observed to increase hepatic and splanchnic blood flow in a biphasic manner. Blood flow is initially reduced because of sympathetic stimulation and is then increased secondary to the direct vasodilator effect of CO2.67 The heterogeneity, however, observed in the individual changes of splanchnic perfusion secondary to decreased VT supports the concept that the direct local vasodilation of an elevated tissue partial pressure of carbon dioxide (PCO2) is opposed by the increased release of catecholamines in the systemic circulation, with an end result of no significant change.68
Pneumoperitoneum
An association between PPV and pneumoperitoneum has long been described.69,70 The mechanism involves airflow dissection through overdistended alveoli into the pulmonic perivascular sheaths.71 The pocket of air dissects to the mediastinum and migrates through the foramina of Morgagni and Bochdalek to cause free air in the peritoneal space.72,73 Risk factors include high airway pressures, large VT, noncompliant lungs, and preexisting pulmonary disease, including obstructive airway disease and acute respiratory distress syndrome. The diagnosis may be easily mistaken for a perforated viscus. In the absence of leukocytosis, abdominal pain, and peritoneal signs, perforation can be excluded by radiographic imaging with water-soluble contrast material administered via oral, rectal, or enterostomy tube.74,75 Other approaches to define the etiology of pneumoperitoneum include aspiration and analysis of the partial pressure of oxygen (PO2) of the intraperitoneal free gas.76,77 Spontaneous resolution usually occurs within a few days.62,78 On rare occasions, “tension pneumoperitoneum” has been described, in which surgical decompression is necessary, even in the absence of peritoneal signs, to relieve vascular collapse.79
Cardiovascular Complications
Cardiovascular complications are associated with critical illness and mechanical ventilation, but are rarely a direct complication. The most common complications include arrhythmias, myocardial ischemia, usually a type II or demand non–ST-segment elevation myocardial infarction, although primary acute coronary syndrome (ST-segment elevation myocardial infarction) may also occur, as most patients have antecedent risk factors for coronary artery disease.
Most arrhythmias in medical ICU patients are tachyarrhythmia (90%), which are divided almost equally between supraventricular (atrial fibrillation 30%) and ventricular foci (monomorphic VT 49%).80 In surgical ICU patients, 61% of tachyarrhythmias were atrial fibrillation.81 Although arrhythmias may occur as a result of underlying structural heart disease, physiologic alterations associated with the presenting illness and intrathoracic changes associated with PPV may also contribute. Ventilated patients often experience significant hypoxemia and hypercapnia, acidemia or alkalemia, hypokalemia, hypomagnesemia, and hypocalcemia that can all precipitate arrhythmias. Arrhythmias can also be precipitated by many drugs: vasopressors, inotropes, and inhaled β-agonists being the most common offenders. Clinicians must weigh the risk-to-benefit ratio of the drug relative to the degree of arrhythmia: specifically, is the arrhythmia more likely to have been triggered by the bronchospasm, hypoxemia or hypercapnia, or a β-agonist? Impaired electrical conduction of the normal cardiac impulse has obvious systemic effects, leading to decreased cardiac output and compounding the preexisting respiratory compromise. Identification and management of the inciting cause of the arrhythmia is important, as is appropriate management, as directed by advanced cardiac life support guidelines of the American Heart Association.82
Myocardial ischemia in ventilated patients can be primary acute coronary syndrome (plaque rupture) or demand-related ischemia (non–ST-segment elevation myocardial infarction) in patients requiring mechanical ventilation for noncardiac indications. In patients presenting with acute coronary syndrome, the need for mechanical ventilation has been associated with increased mortality rates (as high as 50%).83 Myocardial ischemia can occur at any time during the period of mechanical ventilation, and the increased systemic and myocardial oxygen demand during the time of weaning trials may precipitate acute ischemia.84–88 Diagnosis is complicated by the lack of classical symptoms of chest pain and the low sensitivity of continuous electrocardiogram (ECG) monitoring.89 The diagnosis should be considered in patients failing weaning trials. Diagnostic criteria should include clinical, ECG, and cardiac biomarkers, recognizing the limitations associated with biomarker use in the ICU (troponin elevation in clinical presentations of right-heart strain and elevated pulmonary vascular resistance, false elevations in renal failure). Treatment is based on advanced cardiac life support guidelines,90 including interventional procedures.
Renal Complications
The first report to show the impact of PPV on renal function was published in 1947.91 Since then, several studies have documented an association between mechanical ventilation and development of renal failure in the ICU setting.92–94 PPV increases the odds of developing renal failure threefold when PEEP is below 6 cm H2O and more than 17-fold when PEEP is above 6 cm H2O, despite volume replacement, maintenance of normal filling pressures, and adequate oxygen delivery.92 Various mechanisms have been proposed to explain the alterations in renal function in patients receiving PPV, although three mechanisms are judged dominant: hemodynamic, neurohormonal, and biotrauma (Fig. 41-3).
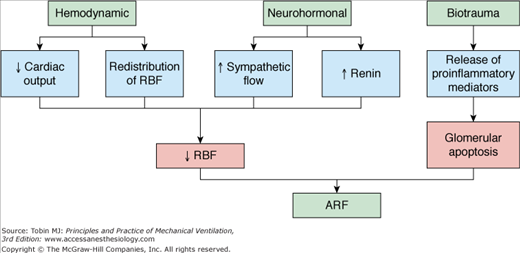
The systemic hemodynamic effects of PPV originate in a complex interaction between intrathoracic pressure, intravascular volume, and cardiac output. The increase in intrathoracic pressure has been shown to correlate with a decrease in renal plasma flow, glomerular filtration rate, and urine output.95,96 The reported effects, however, of PPV on glomerular filtration rate and renal blood flow are variable and may reflect differences in hydration status, underlying pulmonary and cardiac dysfunction, and use of vasoactive agents.97 It has been suggested that redistribution of intrarenal blood flow from the cortical to juxtamedullary nephrons may play an important role in causing reduction of renal function during PEEP.98 This may lead to decreased urinary output and creatinine clearance suggesting that small decreases in renal blood flow can seriously affect renal function.
PPV induces several neurohormonal changes in the sympathetic outflow, the renin–angiotensin axis, nonosmotic vasopressin release, and the atrial natriuretic peptide production. Thus far, no definite correlation between antidiuretic hormone levels99,100 or atrial natriuretic factor,101,102 and renal function during PPV has been confirmed. PPV, however, increases plasma renin levels by twofold,96,102 driven by an increase in sympathetic tone. Consequently, renal blood flow and glomerular filtration are reduced, causing103,104 a drop in distal tubule delivery of sodium. This, in turn, leads to further activation of the renin–angiotensin–aldosterone axis and increased sodium avidity. Clinically, these changes are manifested by a decrease in urine output as a result of decreased osmolar excretion. There are no definitive therapeutic trials for the treatment of PPV-induced renal hypoperfusion. Several small trials have shown that fluid administration and the use of vasoactive drugs (dopamine at 5 μg/kg/min or fenoldopam) may improve renal function,101,105 but results are not conclusive. It is unlikely that approaches focused solely on the hemodynamic and neurohormonal mechanisms of PPV-induced renal dysfunction will be clinically effective in preventing or treating this multifactorial problem.
In addition to altering renal blood flow, PPV can impact renal function through the release of proinflammatory cytokines. These include IL-8, IL-6, and TNF-α, which promote glomerular and tubulointerstitial sequestration of neutrophils, upregulation of leukocyte adhesion molecules, and a decrease in filtration fraction associated with alterations in vascular tone.106–111 Moreover, injurious ventilation strategies, such as high VT and low PEEP, induces glomerular cell apoptosis via a soluble FasL-mediated pathway.107 Regardless of the proposed mechanism, it is clear that derangements in the innate immune/inflammatory response, oxidative stress and cellular necrosis/apoptosis are important components of this organ cross talk in response to injury.
Several authors have described the effects of other ventilation techniques on renal function. Comparison of controlled mechanical ventilation plus PEEP with low-frequency ventilation and extracorporeal carbon dioxide removal resulted in an immediate increase in urinary flow, osmolar clearance, and creatinine clearance.112 Spontaneous breathing during airway pressure release ventilation provides improved systemic blood flow and regional perfusion to the kidneys.113 The effect of permissive hypercapnia on renal function is also well documented: Partial pressure of arterial carbon dioxide (PaCO2) levels correlate inversely with renal blood flow.114 Hypercapnia causes renal vasoconstriction directly115 and stimulates noradrenaline release through activation of the sympathetic nervous system.116 Indirectly, hypercapnia induces systemic vasodilation, which results in a drop of the systemic vascular resistance and subsequent activation of the renin–angiotensin–aldosterone system.117 As a result, renal blood flow becomes compromised and worsen glomerular filtration rate. These hypocapnic changes occur independently of partial pressure of arterial oxygen (PaO2),118 suggesting that PaCO2 plays a pivotal role in determining the vascular response to changes in blood-gas pressures.
Infectious Complications
Most clinical investigations indicate that 25% to 50% of ICU patients experience one or more nosocomial infection.119–121 Fever is the usual trigger for a set of diagnostic and therapeutic interventions. Although the source of fever is not always identified, ventilator-associated infection, sinusitis, catheter-related bacteremias, nosocomial diarrhea, and wound infections account for most infections.122 Two of these complications (i.e., ventilator-associated pneumonia and sinusitis) are discussed in Chapters 46 and 47.
Infection of central venous catheters is a particular problem with a mortality of 10% to 35%.123 The absolute number of catheter-related infections is rising annually because of increasing use, although incidence per catheter days has actually decreased.124 The catheter insertion site itself provides the most direct route of entry for the pathogen and this is the most common cause of infection.123 These infections are caused mainly by gram-positive bacteria, in particular Staphylococcus aureus and coagulase-negative staphylococci such as Staphylococcus epidermidis.125 Infections, however, can be caused by a wide range of microorganisms including Enterococci, Candida spp., Acinetobacter spp., Pseudomonas spp., and Klebsiella spp.126 Catheter management depends on the likely specific pathogens and the individual colonization profile of the patient (Fig. 41-4).126
Figure 41-4
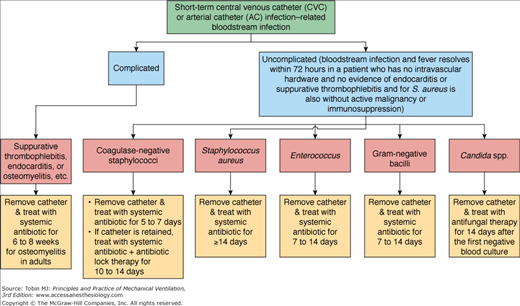
Approach to the management of patients with central venous catheter-related or arterial catheter-related bloodstream infection. (Reprinted with permission from Mermel LA, Allon M, Bouza E, et al. Clinical practice guidelines for the diagnosis and management of intravascular catheter-related infection: 2009 Update by the Infectious Diseases Society of America. Clin Infect Dis. 2009;49:1–45 with permission of Oxford University Press.)
Infectious causes of nosocomial diarrhea include Pseudomonas aeruginosa, Escherichia coli, and cryptosporidium, although C. difficile accounts for the majority of hospital-acquired diarrhea or colitis.127 Prior antibiotics, advanced age, gastric acid suppression, and gastrointestinal surgery or manipulation are major risk factors for symptomatic infection. At least 20% of adults ventilated for more than 1 week are colonized with C. difficile and up to 4% develop infection.128 A hypervirulent strain, the North American pulse-field gel electrophoresis type 1 (NAP1) strain, has been implicated as a cause of C. difficile infection of increasing severity.129,130 A deletion in the toxin regulatory gene, tcdC, is thought to allow this strain to overproduce toxin A and toxin B by as much as 15- to 20-fold.129 Outbreaks caused by this strain are strongly associated with use of fluoroquinolones, although other antibiotics are also implicated.131 For C. difficile infection of every severity, cessation of the inciting antibiotic is the first step in management whenever possible. Metronidazole and vancomycin are the antibiotics most commonly used to treat C. difficile in patients with symptomatic infection. A novel macrocycle antibiotic, OPT-80, is currently in phase 3 trials. OPT-80 is minimally absorbed from the gastrointestinal tract and well tolerated in most subjects.132 Although highly effective against C. difficile, OPT-80 leaves the majority of the gram-negative anaerobic flora of the gastrointestinal tract intact. Another commonly employed strategy is the administration of probiotics. In several randomized controlled trials,133–135 however, the combination of Saccharomyces or Lactobacillus probiotics with conventional antibiotic therapy has failed to show significant benefit.
Hematologic Complications
Ninety-five percent of ventilated patients develop anemia after the third ICU day.136 A significant part is attributed to frequent and excessive blood draws. ICU patients are phlebotomized 3.4 times a day (mean volume of 41.5 mL of blood) as compared to 1.1 times a day for patients on a general ward.137 Occult gastrointestinal bleeding represents another source of blood loss. The condition is aggravated by coagulation disorders that include platelet dysfunction, thrombocytopenia, loss of coagulation factors, and endothelium-related coagulation disorders.138 Several investigations in the last decade, however, suggest that anemia in ICU patients bears many similarities to that associated with chronic disease. More than 90% of ICU patients have low serum iron, total iron-binding capacity, and iron-to-total-iron-binding-capacity ratio, but have a normal or, more usually an elevated, serum ferritin level.139,140 At a time when the iron studies are abnormal, serum erythropoietin levels are only mildly elevated with little evidence of reticulocyte response to endogenous erythropoietin.139 Various proinflammatory cytokines are implicated in this blunted response. Inhibition of renal erythropoietin-gene expression in rat kidneys has been demonstrated for IL-1 and TNF-α.141 In some cases, a shorter erythrocyte life span may aggravate the anemia. There are strong indications that the mononuclear phagocytic system plays a key role, with enhanced iron entrapment and a temporary inhibition of iron release,142 while the bone marrow is still capable of incorporating iron and of reacting to exogenous erythropoietin.
Critically ill patients respond to treatment with recombinant human erythropoietin and parenteral iron with increased erythropoiesis.143 Despite early promising results,144,145 two large placebo-controlled trials investigating the exogenous erythropoietin in critically ill patients failed to show improved 28-day mortality, shorter duration of mechanical ventilation, decreased ICU length of stay, or reduction in red-cell transfusions.143,146 There was, however, an increased propensity for thrombotic vascular events in the erythropoietin arm.146
The incidence of thrombocytopenia in ICU patients varies from 23% to 41%, depending on the definition of thrombocytopenia and the ICU type.147–149 Thrombocytopenia is often multifactorial (Table 41-2) and a marker of disease severity.150 Nearly all studies have found an inverse correlation between platelet count and prolonged ICU stay or hospital mortality.151–153 The magnitude of the drop in platelet count correlates more closely with adverse outcomes than the nadir in absolute count. The platelet count pattern over time provides important clues as to the cause of the thrombocytopenia. A gradual decrease in platelet counts over several days suggests worsening of the underlying disease and is often associated with multiorgan failure, whereas a new, rapid decrease in platelet count that begins after the fourth day is typical for immune-mediated causes. A classic example is heparin-induced thrombocytopenia, which is frequently considered as a cause of thrombocytopenia in ICU patients, although its incidence is only 0.3% to 0.5%.154,155 It occurs in two forms. With the more common, type I, form, which occurs in 10% to 20% of patients receiving unfractionated heparin, nonimmune mechanisms produce a drop in platelet counts 1 to 4 days after the onset of therapy. The type I form is not associated with hemorrhagic or thrombotic sequelae, and management involves observation; platelet counts normalize in most patients despite heparin continuation.156 With the type II form, in contrast, 30% to 80% of patients experience thrombotic sequelae. Venous thromboses outnumber arterial thromboses by 4:1, and life-threatening pulmonary embolism occurs in 25% of patients who have thrombosis.156 The type II form occurs in 1% to 3% of patients who receive unfractionated heparin, and is induced by antibodies of the immunoglobulin G class against platelet factor 4-heparin complexes.156 Heparin-induced thrombocytopenia is suspected when a platelet count falls by more than 50% after 4 days of treatment, and can present with new thrombosis, typically occurring 5 to 14 days after the start of prophylactic or therapeutic doses of heparin. To prevent new thrombosis, nonheparin anticoagulant therapy is required. Four drugs are approved for this purpose: (a) two direct thrombin inhibitors, lepirudin and argatroban; (b) a heparinoid, danaparoid; and (c) an antifactor Xa inhibitor, fondaparinux.
Related posts:







Full access? Get Clinical Tree


