CHAPTER 16
Common Endocrine Disorders: Diseases of the Adrenal, Thyroid, and Parathyroid Glands
Margaret Donat, MD
THE ADRENAL GLANDS
The adrenal glands are very small organs with complex functions. These glands sit on top of the kidneys and consist of a medulla (the center) that produces catecholamines, namely, epinephrine and norepinephrine, surrounded by a cortex that secretes glucocorticoids and mineralocorticoids. Diseases of the adrenal glands can lead to syndromes of wasting, hypotension, and hypoglycemia if adrenal hormones are lacking, whereas diseases that increase the production of adrenal hormones can produce hypertension, weight gain, and virilization. The following section gives the primary care provider information needed for the diagnosis and management of several adrenal diseases: adrenal insufficiency, adrenal overactivity, hyperaldosteronism, hypoaldosteronism, and pheochromocytoma. Appropriate referral information is also given.
Anatomy, Physiology, and Pathology
The adrenal glands are small pyramidal organs, measuring 3 × 6 cm and weighing about 4 to 6 g each, embedded in the retroperitoneal fat medial to the superior pole of the kidneys. The blood supply branches off multiple sites; vascular sites drain from the suprarenal vein into the inferior vena cava on the right and the renal vein on the left. Autonomic innervation of the glands is extensive.
Histologically, the cortex is composed of three layers: the zona glomerulosa, which secretes mineralocorticoids such as aldosterone (which has an important role in the maintenance of salt balance and blood pressure regulation in the body); the zona fasciculata, which secretes glucocorticoids such as cortisol; and the zona reticularis that secretes androgens, such as dihydroepiandrosterone (DHEA), DHEA-sulfat (DHEA-S), and the precursor to testosterone. The medulla arises from neural crest cells. It is very vascular and consists of chromaffin cells, arranged in a network, which secrete catecholamines (Taub & Wolford, 2009).
When adrenal loss is caused by autoimmune disease, antibodies to all three zones of the cortices of the adrenal glands are found in 60% to 75% of patients, causing a decrease in size. Normal adrenal architecture is replaced by fibrosis. When adrenal failure is caused by tuberculosis or fungal infection, the glands are often large and calcified. Adrenal insufficiency can also occur secondary to hemorrhage within the gland, which has a plentiful arterial supply but limited venous drainage, increasing the risk of thrombosis (Falorni et al., 2011).
Synthesis of the steroid hormones is regulated by the hypothalamic–pituitary–adrenal axis (HPA) and, with the exception of aldosterone from the zona glomerulosa, is dependent on adrenocorticotrophic hormone (ACTH). ACTH is produced in the anterior pituitary. Its regulation is maintained by the feedback inhibition of cortisol. In addition, corticotropin-releasing hormone (CRH) stimulates ACTH release. Circadian rhythms and stress also influence the release of ACTH. Cortisol, when present in higher-than-physiological levels, will inhibit ACTH secretion (negative feedback loop). ACTH and cortisol have a diurnal variation, with maximal secretion between 2 a.m. and 8 a.m. and nadir around midnight. ACTH secretion is also increased by fever, hypoglycemia, pregnancy, strenuous exercise, anorexia nervosa, and depression.
Corticotropin (ACTH) can stimulate aldosterone secretion, but its effect is short lived. Aldosterone is instead regulated by the renin–angiotensin system and potassium. Receptors in the wall of the afferent arteriole of the kidney respond to inadequate vascular volume and stimulate the release of renin from the juxtaglomerular cells. Renin cleaves angiotensinogen, a protein made by the liver and found in the plasma, to angiotensin I, which in turn is converted to angiotensin II primarily in the lungs by angiotensin-converting enzyme (ACE), which is elaborated by pulmonary endothelial cells, and cleaves angiotensin I, a decapeptide, to angiotensin II, an octapeptide. Angiotensin II binds to a membrane receptor on the zona glomerulosa cells that is in the family of G-protein-type receptors. Activated second messengers raise intracellular calcium concentrations, leading to aldosterone secretion. Potassium can also increase aldosterone secretion via depolarization of cell membranes, leading to calcium influx through voltage-gated channels (Taub & Wolford, 2009).
The primary precursor of adrenal steroid hormones is cholesterol, which is stored in the adrenal cortex. Multiple enzymatic steps produce the steroid hormones along three pathways, producing mineralocorticoids, glucocorticoids, and androgens.
 ADRENAL INSUFFICIENCY
ADRENAL INSUFFICIENCY
Adrenal insufficiency results from a decreased concentration of glucocorticoids, such as cortisol. The most common cause is the withdrawal of exogenous steroids, often given to treat asthma or rheumatologic conditions. Primary adrenal insufficiency is caused by loss of the adrenal cortex; more than 90% of the cortex must be destroyed for adrenal insufficiency to occur. Secondary adrenal insufficiency is the loss of cortisol secretion because of loss of pituitary ACTH secretion. Causes of adrenal insufficiency are presented in Table 16.1.
Epidemiology
In industrialized nations, primary adrenal insufficiency is usually caused by autoimmune destruction of the adrenal cortex. About 70% to 90% of the cases of adrenal loss involve autoimmune adrenal insufficiency or Addison’s disease. This can occur from age 17 to 72 years, but usually presents by age 40 years. Polyglandular autoimmune syndrome occurs mostly in women (70%); those with isolated autoimmune adrenal insufficiency are mostly male (71%) if they are in the first two decades of life. In the third decade, rates are equal for male and female; thereafter, patients are predominantly female (81%), for unknown reason. Half of the patients with autoimmune adrenal insufficiency have other autoimmune illnesses, such as early gonadal failure, insulin-dependent diabetes mellitus, Hashimoto’s hypothyroidism, Graves’ disease, hypoparathyroidism, or pernicious anemia. Schmidt’s syndrome is the combination of autoimmune adrenal insufficiency and diabetes with or without hypothyroidism. When more than two autoimmune diseases coexist, the syndrome is called polyglandular autoimmune syndrome, where antibodies to 21-hydroxylase (an enzyme involved in steroid synthesis) are found in the blood.
The second leading cause of adrenal insufficiency, found in 7% to 20% of those with adrenal loss, is cortical destruction from infectious diseases, such as tuberculosis or fungal infections. The cortical loss can be caused by metastatic cancer or lymphoma, adrenal hemorrhage or infarction, or drugs. In developing countries, tuberculosis rather than autoimmune disease is the leading cause of adrenal insufficiency. Tuberculosis can destroy the adrenal glands and replace its tissue with caseous nodules and fibrosis; disseminated fungal infections such as histoplasmosis, paracoccidioidomycosis, blastomycosis, cryptococcosis, and coccidioidomycosis can also destroy the glands and cause adrenal insufficiency. Adrenal failure has decreased in HIV patients because of widespread use of antiretroviral therapy (Chakera & Vaidya, 2010; Erichsen et al., 2009).
Causes of Adrenal Insufficiency |
ETIOLOGY | OCCURRENCE |
Primary Adrenal Insufficiency | |
Autoimmune Tuberculosis (#1 infectious cause worldwide) Others Fungal infections Adrenal hemorrhage Congenital adrenal hyperplasia Sarcoidosis Amyloidosis HIV infection (most common infectious cause in the United States) Syphilis African trypanosomiasis Hemorrhagic infarction Adrenoleukodystrophy Adrenomyeloneuropathy Metastatic disease Drugs (by inhibiting cortisol synthesis) | 70%–90% 7%–20% 10% |
Secondary Adrenal Insufficiency | |
Exogenous steroid use After cure of Cushing’s Pituitary and hypothalamic lesions | Very common Common Uncommon |
Source: Nieman (2014).
Other causes of adrenal failure include hemorrhage in the adrenal gland causing infarction (Rosenberger et al., 2011). Unrecognized, this can lead to shock and death, which is then incorrectly attributed to sepsis or ischemia. This is seen in patients who have hypercoagulable states such as lupus, diabetes, or pregnancy; patients using anticoagulants; or patients in the postoperative period. This can also be seen during sepsis, particularly that caused by meningococcemia, pneumococcal pneumonia, Escherichia coli, Neisseria gonorrhea, Staphylococcus aureus, or Haemophilus influenzae (Waterhouse–Friderichsen syndrome). Patients with antiphospholipid syndrome, often associated with lupus, can also lose adrenal function because of arterial and venous thrombi (Bermas, Erkan, & Schur, 2014).
Drugs that work by interfering with steroid synthesis or that accelerate steroid degradation can cause loss of adrenal function. These drugs include those used to treat excessive steroid production (i.e., Cushing’s syndrome) and adrenal carcinoma, such as aminoglutethimide, mitotane (o, p’-DDD, related to the insecticide DDT), and ketoconazole. The latter, for example, inhibits two enzymes in the glucocorticoid-synthetic pathway and binds to the glucocorticoid receptor. Drugs such as rifampin, phenytoin, and phenobarbital accelerate the catabolism of cortisone by stimulating hepatic microsomal enzymes. Their use causes adrenal insufficiency in patients with compromised adrenal function or limited pituitary reserve (Fleseriu et al., 2013; Gordijn, Gemke, van Dalen, Rotteveel, & Kaspers, 2012).
Less-common causes of adrenal cortical loss include metastatic disease, although adrenal failure is rare. This can occur in up to 40% to 60% of women with breast or lung cancer, 30% of those with malignant melanoma, and 14% to 20% of patients with gastric or colon cancer. Other infiltrating diseases, such as amyloidosis, sarcoidosis, or hemochromatosis, can lead to loss of adrenal cortical function. Familial disorders, including X-linked or autosomal recessive metabolic disorders, adrenoleukodystrophy, and the milder adrenomyeloneuropathy, all cause adrenal deficiency; these disorders primarily affect 1 in 20,000 males (Moser, Raymond, & Dubey, 2005).
History and Physical Examination
Adrenal insufficiency can present as an insidious process or as an acute crisis (Salvatori, 2005; Toogood & Stewart, 2008). Patients complain of anorexia, weight loss, and weakness (see Table 16.2). Fatigue, sweating, and loss of concentration can occur, especially as the loss of glucocorticoids enhances hypoglycemia. About half the patients have gastrointestinal complaints, usually nausea, vomiting, abdominal or loin pain, and diarrhea. In primary adrenal insufficiency, the patient notes darkening of the skin, caused by enhanced secretion of proopiomelanocortin-derived peptides. The increased pigmentation is found especially on the face, at the elbows, knees, creases of the palmar surface, gingival margin, or buccal mucosa; vitiligo is also seen. Hypotension, with systolic readings <110 mmHg, and orthostatic blood pressure changes, is found. Women report hair thinning and irregular menses. Psychiatric symptoms are seen in 64% to 84% of patients with adrenal insufficiency. The most common findings are depression, apathy, or confusion, but there are reports of psychosis, paranoia, schizophrenia, and self-mutilation (Chakera & Vaidya, 2010; Marik et al., 2008).
Symptoms and Signs of Adrenal Insufficiency |
ACUTE |
Abdominal pain, postural hypotension, fever, weakness, confusion |
CHRONIC |
Weakness, fatigue, salt craving, anorexia, nausea, vomiting, diarrhea, apathy, dizziness, and syncope; weight loss, orthostatic hypotension, vitiligo, pigmentation of skin and mucous membranes, and alopecia |
Adrenal hemorrhage with loss of adrenal function can be a complication of several illnesses, including sepsis, trauma with shock, coagulopathies, and ischemic disorders. The clinical signs can be vague, but usually patients have abdominal, flank, back, or chest pain with fever and hypotension. Anorexia, vomiting, psychiatric symptoms, and abdominal rigidity with rebound also occur (Chakera & Vaidya, 2010; Marik et al., 2008).
Pituitary or hypothalamic disease can lead to secondary adrenal failure. Secondary adrenal failure is often associated with other secondary losses, such as hypothyroidism or hypogonadism (Reimondo, Bovio, Allasino, Terzolo, & Angeli, 2008b). Thus, patients may present with fatigue, loss of menses, loss of libido, or difficulty getting erections. In secondary adrenal insufficiency, aldosterone secretion is preserved because aldosterone is regulated by the renin–angiotensin axis rather than ACTH (Marik et al., 2008).
Loss of aldosterone secretion can occur in isolation (hyperreninemic hypoaldosteronism) or as a result of diminished secretion of renin or angiotensin II (hyporeninemic hypoaldosteronism). Hyporeninemic hypoaldosteronism is commonly found in patients with mild renal insufficiency (e.g., diabetes [50% of patients]) or those with nephritis but it can also be caused by use of nonsteroidal anti-inflammatory drugs (NSAIDs; see section on hypoaldosteronism). Hyperreninemic hypoaldosteronism is usually seen in critically ill patients and may be caused by hypotensive injury to the adrenal glands. It can also be seen in patients with diabetes or those taking heparin (Marik et al., 2008). These patients are usually asymptomatic but can have cardiac arrhythmias or muscle weakness because of hyperkalemia (Karet, 2009).
Diagnostic Studies
LABORATORY FINDINGS
Fifty to sixty percent of patients with primary adrenal insufficiency are hyperkalemic, and 90% are hyponatremic from increased release of antidiuretic hormone (ADH) causing renal salt wasting and water retention. In primary but not secondary adrenal insufficiency, mineralocorticoids (aldosterone) are lost along with the glucocorticoid secretion, causing intravascular volume depletion (hypotension) and elevated serum potassium levels. Primary adrenal insufficiency frequently causes hypoglycemia, hypercalcemia, and a mild normocytic anemia with lymphocytosis and mild eosinophilia (Mount, 2009).
In secondary adrenal insufficiency, hyponatremia but not hyperkalemia can occur, but from a different cause. The lack of ACTH leads to low cortisol levels, which increases vasopressin secretion and water retention. These patients also show evidence of other pituitary hormone abnormalities, such as elevated prolactin if there is a pituitary tumor, or low levels of thyroid-stimulating hormone (TSH), follicle-stimulating hormone, luteinizing hormone, and free thyroxine.
In adrenal hemorrhage, there is a precipitous drop in the hematocrit while the number of white blood cells increases (leukocytosis; Baker et al., 2012). Hyponatremia and hyperkalemia with renal insufficiency (azotemia) and acidosis are also seen.
The lack of response of the adrenal gland to ACTH stimulation may help confirm the diagnosis of adrenal insufficiency. Random cortisol levels may not distinguish patients with mild disease from the normal population, but morning cortisol levels >25 mcg/dL (>525 nmol/L) are likely to indicate normal function, whereas patients with 8 to 9 a.m. cortisol levels <4 mcg/dL (83 nmol/L) probably have adrenal insufficiency. The ACTH stimulation test can be done at any time and consists of giving 250 mcg (85 nmL, or 40 international units) of synthetic ACTH (cosyntropin) intravenously or intramuscularly. To differentiate primary from secondary disease, an ACTH level is drawn 30 minutes before the synthetic ACTH is given and 60 minutes after the injection; the minimum value is 18 to 20 mcg/dL or 500 to 550 nmol/L (before or after the injection). Measurement of ACTH in the basal sample will be high in primary disease (>100 pg/mL or 22 pmol/L) and low in secondary disease. Alternatively, serum aldosterone could be measured; if low, it is indicative of primary disease, whereas its level is normal in secondary disease (Deutschbein, Unger, Mann, & Petersenn, 2009).
IMAGING STUDIES
In patients with primary adrenal insufficiency from autoimmune disease, imaging of the adrenal glands is not necessary. In other cases, CT of the adrenal glands should be done to diagnose fungal disease or cancer. In patients with secondary disease from a pituitary or hypothalamic process, MRI of the pituitary and hypothalamic regions is done.
Treatment Options, Expected Outcomes, and Comprehensive Management
The usual replacement is cortisone, 12 to 15 mg/m2/d, usually given as 25 mg in the morning and 12.5 mg in the evening to try to mimic the natural circadian rhythm of higher steroid production in the morning (Nieman, 2013). If other steroids are used, dosages are adjusted according to the relative potency of the drug. For example, prednisone is about four times as potent as hydrocortisone, so replacement is given as 5 mg in the morning and 2.5 mg in the evening. Because steroids can cause gastritis, weight gain, and osteoporosis, the goal is to give the lowest dosage that relieves symptoms; this can be as low as 15 to 20 mg/d of hydrocortisone. The steroid replacement dose may be adjusted based on 24-hour measurements of urinary cortisol. Mineralocorticoids are necessary in 75% of patients with primary adrenal insufficiency to prevent sodium loss, intravascular volume depletion, and hyperkalemia; the dosage is usually 50 to 200 mcg 9-alpha-fludrocortisol (Florinef) given each morning with a liberal-salt diet. This aldosterone substitute can be adjusted by measuring blood pressure, potassium, and plasma renin activity (PRA), which should be in the upper end of the normal range (Fitzgerald, 2008).
Teaching and Self-Care
Concern about causing iatrogenic alterations in the pituitary–adrenal axis is widespread because of the common use of glucocorticoids to treat chronic diseases such as arthritis, inflammatory bowel disease, asthma, and emphysema.
![]() CLINICAL WARNING:
CLINICAL WARNING:
When the adrenal axis is suppressed from glucocorticoid use, abruptly decreasing or stopping the medication or missing a dose could precipitate an adrenal crisis (adrenal insufficiency) with symptoms of weakness, fever, nausea, vomiting, abdominal pain, confusion, hypotension, hypoglycemia, and loss of consciousness. This is life threatening and requires emergency medical treatment.
The degree of suppression of the adrenal axis depends on many factors, such as the type of steroid used, the dosage, and the duration of treatment. Nonetheless, many studies show that suppression is not predictable in individual patients, and thus random cortisol levels are not helpful. Patients taking 20 mg or more of prednisone (or its equivalent) for more than 5 days should be suspected of having adrenal suppression, and thus are at risk of adrenal crisis if the medication is not appropriately weaned. Some patients have symptoms of adrenal insufficiency with normal cortisol and ACTH levels, probably from a sudden elevation in prostaglandin levels after steroid disuse (steroid withdrawal syndrome).
Alternate-day therapy is an approach used by many providers to minimize adrenal axis suppression and decrease side effects from glucocorticoids while sustaining therapeutic effects. This use of a short-acting steroid (prednisone or methylprednisolone) every 48 hours should be the regimen of choice in patients facing more than a few weeks of glucocorticoid therapy for conditions other than adrenal insufficiency. Those lacking cortisol or ACTH secretion must take replacement daily.
During periods of stress such as febrile illness (temperature >100.4°F), gastroenteritis, or outpatient surgery with substantial blood loss (e.g., tooth extraction), signs and symptoms of adrenal crisis, or unconsciousness, additional steroid coverage is given. The oral dose of steroid can be doubled for the first 2 or 3 days until the underlying fever or “24-hour bug” resolves. If patients are unable to take medications orally, they can be instructed to give themselves glucocorticoid by subcutaneous injection or to use glucocorticoid suppositories for replacement. Patients may also make changes in their diet and lifestyle to manage symptoms of adrenal insufficiency. These include eating a healthy diet that contains ample fresh fruits and vegetables and is low in simple carbohydrates. Moderate exercise should be encouraged, both for overall good health and to aid in stress management. If the latter is a problem, patients may benefit from stress management techniques that are individualized for them. Patients on steroid replacement should be encouraged to wear a Medic-Alert bracelet or to carry a medical identification care stating that they use this medication. Patient should always carry a syringe with a vial of dexamethasone (glucocorticoid) for safety. Typically, an injection is given into the thigh muscle.
Because steroid-related gastritis can be a problem, patients should be encouraged to avoid smoking and the use of aspirin and ibuprofen and to limit caffeine and alcohol intake. They should be advised to take their steroid with food. Some patients may find that eating small, frequent meals helps their tolerance of steroid medication.
Self-care for bone loss and osteoporosis is covered in another chapter.
![]() CLINICAL WARNING:
CLINICAL WARNING:
Chronic adrenal failure may persist for months or years before diagnosis, but acute adrenal insufficiency (adrenal crisis) must be promptly recognized and treated to avoid death, which is usually caused by the underlying illnesses. It can be difficult to diagnose acute adrenal crisis; it is often misdiagnosed as sepsis or an acute abdomen. Acute loss of cortical secretion leads to tachycardia, hypotension, and vascular collapse, severe abdominal pain, nausea and vomiting, hyponatremia, and hyperkalemia.
Clinical Pearls
 The provider should consider referring a patient with suspected adrenal insufficiency to an endocrine specialty practice for diagnosis and management planning.
The provider should consider referring a patient with suspected adrenal insufficiency to an endocrine specialty practice for diagnosis and management planning.
![]() CLINICAL WARNING:
CLINICAL WARNING:
Acutely ill patients may be severely hypotensive. They may need emergency referral.
When a patient with known adrenal insufficiency must undergo emergency surgery, steroid coverage is increased and is given intravenously until oral medication can be reliably taken.
If patients have a history of steroid use, and in whom the length of steroid treatment, time since last use, or the dosage that was given is not clearly known, the safest and most practical clinical solution is to assume that they may be adrenal-insufficient.
![]() CLINICAL WARNING:
CLINICAL WARNING:
It is a recommended practice to give steroid coverage during emergency situations for any patient taking steroids over the past 12 months, even if the patient took alternate-day glucocorticoids.
Additional Resources for Patients
 National Library of Medicine: www.nlm.nih.gov/medlineplus/healthtopics.html
National Library of Medicine: www.nlm.nih.gov/medlineplus/healthtopics.html
 National Adrenal Disease Foundation: www.nadf.us
National Adrenal Disease Foundation: www.nadf.us
 Hormone Health Network: www.hormone.org
Hormone Health Network: www.hormone.org
 National Institute of Diabetes and Digestive and Kidney Diseases: www.niddk.nih.gov
National Institute of Diabetes and Digestive and Kidney Diseases: www.niddk.nih.gov
 ADRENAL OVERACTIVITY
ADRENAL OVERACTIVITY
Cushing’s Disease and Cushing’s Syndrome
Cushing’s syndrome is a group of diseases characterized by excessive glucocorticoid secretion. The syndrome can be caused by increased ACTH secretion (pituitary or ectopic) or it can be ACTH independent (adrenal disease). In Cushing’s disease, a pituitary lesion leads to increased ACTH secretion, which in turn leads to bilateral adrenal hyperplasia and increased glucocorticoid production.
EPIDEMIOLOGY
The most common cause of Cushing’s syndrome is iatrogenic, from administration of excessive glucocorticoids (oral, parenteral, or topical). Endogenous excessive glucocorticoid production causing Cushing’s syndrome is most often ACTH-dependent. A pituitary tumor secreting excessive ACTH is the cause in 68% of all patients; this is called Cushing’s disease. Ectopic production of ACTH is caused by numerous types of tumors, like small cell lung cancer, bronchial carcinoma, thymic and pancreatic carcinoids and so on; this occurs in 12% of cases and is also ACTH dependent. ACTH-independent disease is caused by autonomous adrenal activity (adrenal adenomas or carcinomas) in 19% of cases. Cushing’s syndrome occurs at a rate of 10 cases per 1 million in the population.
HISTORY AND PHYSICAL EXAMINATION
Cushing’s syndrome should be considered in patients who have a history of hyperglycemia, hypertension, osteoporosis, and a recent gain in weight without use of steroids. The presence of marked virilization in women increases the likelihood of adrenal carcinoma, whereas marked hypertension may indicate ectopic ACTH secretion. The symptoms and signs of hypercortisolism are caused by alterations in the metabolism of lipids, protein, and carbohydrates. Signs and symptoms include:
 Central obesity
Central obesity
 Hypertension
Hypertension
 Glucose intolerance or diabetes
Glucose intolerance or diabetes
 Facial rounding and plethora
Facial rounding and plethora
 Filling in of supraclavicular fat pads and central obesity
Filling in of supraclavicular fat pads and central obesity
 Loss of muscle strength and thinning of arms and legs
Loss of muscle strength and thinning of arms and legs
 Easy bruising
Easy bruising
 Acne
Acne
 Hirsutism
Hirsutism
 Irregular menses in women; decreased libido and gynecomastia in men
Irregular menses in women; decreased libido and gynecomastia in men
 Deepening voice
Deepening voice
 Peripheral edema
Peripheral edema
 Loss of height, back pain, and osteoporosis
Loss of height, back pain, and osteoporosis
Fatigue, weakness, irritability, polyuria, polydipsia, and increased frequency of skin and urinary tract infections from hyperglycemia also are common complaints. Altered protein metabolism can lead to easy bruising and striae wider than 1 cm on the abdomen or proximal extremities. Oligomenorrhea or amenorrhea occurs in premenopausal women; men report decreased libido (Toogood & Stewart, 2008). Depression and insomnia often occur; major depression is found in as many as 66% to 75% of patients.
The typical cushingoid appearance is that of a patient with a round face (moon facies) with plethora (telangiectasias), fat pads that bulge above the supraclavicular fossae, and central obesity with spindle-like extremities. An increased prominence of the dorsal fat pad (buffalo hump) is less specific for Cushing’s syndrome and can be found in patients with weight gain from any cause. There may be increased facial hair, thinning of the skin with petechiae and ecchymosis, and increased pigmentation of the skin, especially in the groin and palmar creases if the Cushing’s syndrome is ACTH dependent. There is proximal muscle wasting from protein catabolism, so the patient has difficulty rising from the seated position. There can also be osteoporosis or frank fractures with minimal trauma.
Pseudo-Cushing’s syndrome is a diagnosis given to the disorder in which patients appear to have the physical signs of Cushing’s syndrome, but the excess glucocorticoid is from secondary causes, such as alcoholism or depression. Patients with alcoholism have hormonal abnormalities that resolve with abstinence. Their hormonal abnormalities are probably caused by increased CRH or impaired hepatic metabolism of cortisol. Major depression can also lead to abnormally regulated cortisol metabolism, although hypersecretion of cortisol is minimal. The hormonal defect disappears on remission of the depression.
DIAGNOSTIC STUDIES
Laboratory Findings • Single cortisol values are not of value in diagnosing Cushing’s syndrome. There is, nonetheless, a loss of the usual diurnal variation in cortisol secretion and evening cortisol levels are equal to or higher than morning values. In a patient with the signs of Cushing’s syndrome, low cortisol values may indicate the previously unsuspected use of exogenous steroids.
Screening tests for Cushing’s syndrome involve either the collection of two or three 24-hour urine samples for free cortisol measurement, or the overnight dexamethasone suppression test, because ACTH secretion is episodic in Cushing’s syndrome. To verify the completeness of a 24-hour urine collection, urine creatinine is also measured in the sample, as its level should not vary by more than 10%. Both tests can have false-negative and false-positive results, but a morning serum cortisol >5 mcg/dL after an 11 p.m. dose of 1 mg dexamethasone or a urine-free cortisol value >80 mcg/dL warrants referral for further investigation. Stress can lead to false-positive results. Testing is best done in the outpatient setting.
The ideal time to screen a patient for Cushing’s syndrome is between midnight and 2 a.m., when cortisol and ACTH levels are usually low because of circadian rhythms. For patient convenience, this can be done after 4 p.m., and an ACTH level >15 pg/mL (>3.3 pmol/L) in the face of a cortisol level >15 mcg/dL (>415 nmol/L) suggests ACTH-dependent Cushing’s, either Cushing’s disease or ectopic ACTH syndrome. If ACTH levels are under 5 pg/mL (1.1 pmol/L) with elevated cortisol levels, a primary adrenal lesion may be present.
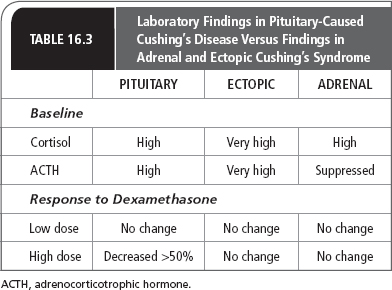
The classic endocrine specialty workup for Cushing’s syndrome involves looking at the endogenous steroid production after low- and high-dose steroids are given. The low-dose test consists of giving 0.5 mg dexamethasone every 6 hours for 48 hours. Because the classic 2-day low-dose and high-dose dexamethasone testing may be too complicated for patients to do in the outpatient setting, some endocrinologists do a more rapid high- and low-dose test. Patients can take increasing amounts of dexamethasone at night and have morning cortisol and ACTH levels measured. For the low-dose test, 2 mg of dexamethasone are given, and for the high-dose test, 8 mg of dexamethasone are given. Table 16.3 presents laboratory findings in pituitary-caused Cushing’s syndrome versus those in adrenal and ectopic syndrome.
A CRH stimulation test is given intravenously. Within 45 minutes of administration, most patients with Cushing’s disease will have an increased level of ACTH and cortisol. Patients with adrenal tumors will not respond because of increased urinary free cortisol and ectopic ACTH secretion; pituitary ACTH is suppressed. A response to CRH thus helps differentiate Cushing’s disease from all other causes of Cushing’s syndrome.
Petrosal venous sinus catheterization is the most direct way of showing pituitary ACTH hypersecretion by revealing a central-to-peripheral ACTH gradient in the blood draining the tumor. Catheters are inserted through the femoral or jugular veins into both inferior petrosal veins. ACTH is measured in petrosal and peripheral venous blood before and within 10 minutes of administering CRH. A central-to-peripheral blood ACTH plasma gradient of ≥2.0 before administering CRH or ≥3.0 after administering CRH confirms a pituitary source of ACTH.
Pseudo-Cushing’s syndrome can be differentiated from Cushing’s syndrome by checking evening cortisol levels. Cortisol concentrations in the blood have a diurnal rhythm, high in the morning and low between 10 p.m. and 2 a.m. This evening nadir is preserved in pseudo-Cushing’s syndrome and not in Cushing’s syndrome, so patients with the fomer should demonstrate a low midnight cortisol value (<5 mcg/dL or 138 nmol/L).
IMAGING STUDIES
Before the advent of MRI, pituitary tumors were identified in <50% of those with Cushing’s disease, because Cushing’s disease is often caused by a microadenoma (a lesion <1 cm). Now, with MRI scanning, pituitary tumors can be identified in up to 75% of those with Cushing’s disease. In some patients, there are signs that the defect originates in the hypothalamus rather than in the pituitary.
Patients with adrenal adenomas have an equal chance of having benign or malignant tumors. MRI imaging, although it is more expensive than CT, can occasionally differentiate benign from malignant masses. With the advent of better CT and MRI scanners, small adrenal lesions (<0.5 cm) are being identified that are nonfunctional; these are called adrenal incidentalomas. Seventy percent to 94% of adrenal masses found incidentally are nonsecretory and benign adenomas (Musella et al., 2013). Minimally invasive or laparoscopic adrenalectomy is now the procedure of choice for benign appearing adrenal tumors ≤6 cm in diameter and weighing <100 g (Conzo et al., 2013). A cancerous lesion is suspected if the tumor is >6 cm.
If no pituitary tumor is seen on MRI and the patient has non-suppressible cortisol secretion, bilateral inferior petrosal vein catheterization and sampling for ACTH during stimulation with CRH can differentiate pituitary from ectopic ACTH-dependent disease. This catheterization procedure is expensive and invasive and can produce dire complications, including brainstem damage; thus, it cannot be recommended as a routine procedure.
![]() CLINICAL WARNING:
CLINICAL WARNING:
Functioning adrenal masses that oversecrete hormone require surgical removal or medical therapy if the former is not possible. CT and MRI scanning cannot distinguish hormonally active from inactive lesions, and lesions that produce hormones can be clinically silent. Most malignant tumors produce hormones, but they can be inefficient at making cortisol. Women with malignant adrenal tumors often appear virilized from overproduction of adrenal androgens, which would provide the clinical clue for further investigation. Men with estrogen-secreting tumors can present with feminization: gynecomastia, testicular atrophy, and decreased libido (Fassnacht, Libé, Kroiss, & Allolio, 2011).
Treatment Options, Expected Outcomes, and Comprehensive Management
SURGICAL TREATMENT
The treatment of Cushing’s syndrome depends on the cause. If Cushing’s disease is found, the pituitary adenoma should be removed by transsphenoidal surgery. The surgery is curative in 60% to 90% of cases.
For larger tumors (macroadenomas), total hypophysectomy is required. If the symptoms of Cushing’s syndrome are very pronounced, bilateral adrenalectomy may be preferred, as this stops the organ damage from hyperglycemia, hypertension, and protein catabolism. In up to 25% of patients treated in this fashion, there is a risk of developing Nelson’s syndrome (a state created by the lack of feedback of exogenous steroid replacement on the pituitary tissue; Barber et al., 2010), because removal of the adrenal glands allows pituitary secretion of ACTH to increase. This is more likely to occur if the original pituitary lesion was an adenoma rather than hyperplasia, and if there was a high level of pretreatment urinary cortisol.
RADIATION THERAPY
Pituitary irradiation can be used, although the cure rate is not high. Relapse of Cushing’s syndrome in adults is less likely when unsuccessful pituitary surgery is followed by radiation.
PHARMACOTHERAPY
In cases of Cushing’s syndrome, where the tumor cannot be resected and radiation may take years to decrease cortisol production, drugs that inhibit steroid production can be used. These include aminoglutethimide and ketoconazole, and a drug that actually destroys adrenal cortical cells, mitotane (o,p’-DDD). Ketoconazole is preferred because of its lack of side effects, but in men, it can inhibit androgen secretion, leading to gynecomastia; in women, it can lead to hirsutism. Aminoglutethimide and mitotane can cause significant gastrointestinal side effects, particularly anorexia and vomiting. These drugs are less effective in the treatment of Cushing’s disease than in other causes of Cushing’s syndrome because the block in steroid synthesis actually increases ACTH secretion, which can ultimately overcome the drug’s ability to block steroid synthesis.
Teaching and Self-Care
Patients with obesity often present to the primary care provider for evaluation of pituitary or adrenal function to determine if a hormonal imbalance has led to difficulty in losing weight. The patient may know a friend or relative with Cushing’s syndrome or may have seen information in the press, on television, or on the Internet about adrenal gland disorders. It can be difficult to separate patients with syndrome X, a genetic tendency for weight gain, hypertension, type 2 diabetes, gout, and (in women) hyperandrogenism (hirsutism), from those with early Cushing’s syndrome. This is especially true if the patient with syndrome X is depressed. Laboratory findings discussed previously and serial examinations can be helpful, along with the recognition that Cushing’s syndrome is rare and obesity, type 2 diabetes, hypertension, and depression are more common.
Hypersecretion of CRH can occur in depression, stress, anorexia nervosa, excessive exercise, panic disorders, chronic alcoholism or drug withdrawal, diabetes mellitus with neuropathy, and central obesity. There may be higher levels of glucocorticoids in hyperthyroidism and premenstrual tension syndrome and in survivors of childhood sexual abuse. Lower levels of CRH or HPA axis dysregulation and “hypoarousal” may be found in patients with hypothyroidism and seasonal depression and chronic fatigue syndromes, including fibromyalgia. The weight gain, depression, and fatigue in Cushing’s syndrome from chronic hypercortisolism might then be attributed to suppression of CRH secretion.
The primary care provider can assist the patient with Cushing’s syndrome by encouraging regular exercise and stress management. Other healthy lifestyle choices have been previously discussed, including diet and tobacco, alcohol, and caffeine management. The provider should also discuss the benefits of limiting refined carbohydrates in the diet.
![]() CLINICAL WARNING:
CLINICAL WARNING:
Patients with adrenal tumors should at least be screened for pheochromocytoma, because 65% of these may have the signal intensity of adrenal metastases on MRI and appear bright. Percutaneous needle biopsy of an unsuspected pheochromocytoma could lead to release of catecholamines, causing hypertensive crisis, retroperitoneal bleeding, and death.
Clinical Pearls
 The use of female hormones can create a falsely elevated serum cortisol value. If results of screening tests for Cushing’s are equivocal, consider whether the use of female hormones may have influenced free hormone levels. In this instance, measure free cortisol in the urine. Alternatively, birth-control pills or female hormones can be stopped for six to eight weeks before testing.
The use of female hormones can create a falsely elevated serum cortisol value. If results of screening tests for Cushing’s are equivocal, consider whether the use of female hormones may have influenced free hormone levels. In this instance, measure free cortisol in the urine. Alternatively, birth-control pills or female hormones can be stopped for six to eight weeks before testing.
 INCREASED PRODUCTION OF MINERALOCORTICOIDS
INCREASED PRODUCTION OF MINERALOCORTICOIDS
Hyperaldosteronism
Excessive production of mineralocorticoids can occur as the result of primary disease (overproduction of aldosterone) or secondary disease from excessive stimulation by the renin–angiotensin system. In both instances, glucocorticoid production is normal.
Primary hyperaldosteronism can be caused by an aldosterone-producing adenoma, bilateral adrenal hyperplasia (sometimes referred to as idiopathic hyperaldosteronism), or aldosterone-producing carcinoma. In bilateral hyperplasia, it is thought that the adrenals may be stimulated by a pituitary or hypothalamic factor, causing the overstimulation of aldosterone secretion (Nieman, 2013).
EPIDEMIOLOGY
Primary aldosteronism is a rare cause of hypertension, accounting for 1% of patients with hypertension. It should be suspected in patients with unexplained hypertension and hypokalemia even with low-dose diuretics. In those patients, the incidence is increased to 50%. It affects women more often than men, and the peak age of diagnosis is between 30 and 50 years. More than 60% of cases are caused by benign adenomas; 30% of patients have idiopathic hyperaldosteronism.
HISTORY AND PHYSICAL EXAMINATION
Patients with hyperaldosteronism may have no specific symptoms, but could have some complaints caused by mild hypokalemia; a low potassium level is found in up to 90% of patients. These complaints include polyuria, polydipsia, and muscle weakness. When the loss of potassium is severe, there may be paresis or tetany. The loss of potassium will also bring out glucose intolerance. Edema is not seen unless there is coexistent congestive heart disease or nephrotic syndrome. Hypertension (blood pressure >140/90 mmHg) is almost always seen in hyperaldosteronism, although there are some rare patients who are normotensive but have hypokalemic alkalosis caused by hyperaldosteronism.
OTHER CAUSES OF HYPERALDOSTERONISM
Some patients with hyperaldosteronism have hypokalemia but no hypertension; this is called Bartter syndrome and is caused by excessive renin production from hyperplasia of the renal juxtaglomerular apparatus. There is also increased excretion of prostaglandins and the hypokalemia responds to treatment with prostaglandin inhibitors such as indomethacin. Nonetheless, potassium supplementation is often used as the sole therapy because of the undesirable long-term side effects of NSAIDs.
Dexamethasone-suppressible hyperaldosteronism is an autosomal dominant disorder with features identical to those of primary aldosteronism, but the excessive aldosterone secretion can be eliminated by giving steroids such as dexamethasone.
Excessive ingestion of licorice can cause hypertension and laboratory findings of mineralocorticoid excess.
Secondary aldosteronism that also leads to hypokalemia is caused by elevated renin levels. Hyperreninemic hyperaldosteronism can occur in malignant hypertension, unilateral renal artery stenosis, and renin-secreting tumors of the kidney’s juxtaglomerular apparatus.
DIAGNOSTIC STUDIES
Laboratory Findings • For an accurate evaluation of the renin–aldosterone axis, many patients have to discontinue drugs known to affect hormones in the pathway. Spironolactone and estrogens should be stopped for 6 weeks. Prostaglandin inhibitors (NSAIDs), calcium channel blockers, ACE inhibitors, sympathomimetics, and adrenergic inhibitors should be withheld for 2 weeks. If patients experience hypertension after discontinuing these drugs, prazosin or other alpha-1 adrenergic inhibitors can be substituted during this period.
Laboratory testing in patients with hyperaldosteronism demonstrates a low potassium level (3–3.5 mEq/L) with alkalosis. The potassium value may be normal if the patient has been eating a low-sodium diet, so these tests should be done after 2 or 3 days of sodium loading (>200 mEq or 120 mM Na). Primary aldosteronism is suspected if there are borderline or low potassium levels, low stimulated PRA, and high aldosterone levels. Aldosterone values are high in the serum (>14 ng/dL), and urinary aldosterone and potassium levels are also high (K >30 mEq). In secondary hyperaldosteronism, the major distinguishing feature is elevation of PRA. In contrast to secondary hyperaldosteronism, the PRA is suppressed in primary aldosteronism and does not increase in response to an upright position (standing for 4 hours) or volume depletion after a 40-mg dose of furosemide.
![]() CLINICAL WARNING:
CLINICAL WARNING:
Many providers prefer to avoid salt loading in patients who are hypertensive and may be prone to congestive heart failure.
An initial evaluation for hyperaldosteronism is to check the plasma aldosterone level and the ratio of plasma aldosterone to PRA before and 90 minutes after administration of captopril (25–50 mg), an ACE inhibitor. This test does not require dietary preparation and does not expose the patient to salt loading. Those with secondary hyperaldosteronism, like normal patients, show a rise in PRA and a fall in aldosterone levels.
Single Adenoma Versus Bilateral Adrenal Hyperplasia. Once the diagnosis of primary aldosteronism is confirmed, it is important to then distinguish between causative factors: either a single adrenal adenoma or bilateral adrenal hyperplasia.
IMAGING STUDIES
CT scanning can distinguish adrenal hyperplasia from a benign adenoma and can also exclude adrenocortical carcinoma. If the CT is normal or reveals bilateral abnormalities or a unilateral abnormality in a patient older than 40 years, then an adrenal venous sampling is recommended to rule out unilateral disease and assess readiness for surgical management if needed. Aldosterone should be measured the day after surgery to assess cure. Potassium and spironolactone should be discontinued and antihypertensive medications decreased.
OTHER STUDIES
Hypokalemia (K <3 mEq/L) can be seen on electrocardiogram tracings as depression of the ST segment and inversion of the T wave. As potassium levels continue to fall, a prominent U wave is seen in the anterior leads.
Treatment Options, Expected Outcomes, and Comprehensive Management
In patients with a benign adenoma, surgical removal of the adrenal gland that contains the adenoma is curative in more than 70% of patients. Surgery is not indicated in idiopathic hyperaldosteronism (bilateral hyperplasia), which is treated with a potassium-sparing diuretic, ACE inhibitors, or calcium channel blockers, which inhibit aldosterone secretion.
Spironolactone (50–200 mg/d) is the most effective agent for hyperaldosteronism that cannot be treated surgically, as it is a competitive antagonist. It can create side effects because of its antiandrogenic activity, leading to decreased libido, breast pain, and gynecomastia in more than 50% of men treated. In women, breast pain and irregular menstrual bleeding can occur. If spironolactone is not tolerated, calcium channel blockers with potassium-sparing diuretics can be used.
Teaching and Self-Care
Hyperaldosteronism is a rare cause of hypertension. Nonetheless, it should be considered as a diagnosis in patients with high blood pressure who have low potassium levels while not taking diuretics, or in those taking diuretics who appear to need greater potassium supplementation than usual.
Potassium can be replaced orally as a liquid or as a slow-release tablet, which is usually preferred because of the unpleasant taste of liquid potassium. The slow-release tablets must be taken with a copious amount of water to avoid gastric irritation. Most patients taking diuretics do not need routine potassium replacement. Patients who have potassium levels <3 mEq/L, those with coexistent cardiac disease (especially those who take cardiac glycosides [digoxin, digitalis]), those with chronic liver disease, and those at risk for diuretic-induced glucose intolerance (diabetes) should take oral potassium while taking commonly used diuretics.
Self-care management includes maintaining a healthy lifestyle. The provider can counsel the patient about diet, including sources of potassium. Exercise as well as stress-relieving activities can be important parts of the self-care plan. Balancing exercise and activities with adequate sleep and quiet recreation can round out a successful self-management regimen.
![]() CLINICAL WARNING:
CLINICAL WARNING:
Although many providers routinely evaluate outpatients for hyperaldosteronism by oral salt loading or short-term intravenous salt loading (2 L normal saline over 4 hours), these tests can lead to serious side effects, primarily congestive heart failure and severe hypertension. Normal patients have aldosterone values of 5 ng/dL or less after saline infusion; those with hyperaldosteronism have values >10 ng/dL. Some authors advise never giving sodium to a hypokalemic patient in whom primary aldosteronism is suspected. Such loading will increase further potassium loss and trigger cardiac arrhythmias.
It is best to replace potassium orally, unless a life-threatening cardiac arrhythmia or vomiting makes this route unfeasible. It is also best to give the replacement slowly to avoid inducing hyperkalemia, ventricular fibrillation, and cardiac standstill.
 HYPOALDOSTERONISM
HYPOALDOSTERONISM
Hypoaldosteronism should be suspected in any patient with persistent hyperkalemia with no kidney failure and no potassium supplement therapy. Hypoaldosteronism can be a primary disease or can occur secondary to low renin levels. Usual causes of hypoaldosteronism include primary adrenal insufficiency, salt-wasting forms of congenital adrenal hyperplasia, or the use of drugs. Primary loss of aldosterone secretion can also be caused by genetic mutations of enzymes in the synthetic pathway for mineralocorticoids. Hyperreninemic hypoaldosteronism can also occur in critically ill patients as a likely response to chronic stress, ischemia in the zona glomerulosa, or a reaction to pyrogenic factors (e.g., interleukin-1, tumor necrosis factor). Secondary hypoaldosteronism from low levels of renin (hyporeninemic hypoaldosteronism) is a common cause of hyperkalemia. It often occurs in patients with mild renal insufficiency, and most of these patients have diabetes mellitus. Drugs known to inhibit aldosterone secretion or interfere with its actions include heparin, cyclosporin A, calcium channel blockers, beta-blockers, NSAIDs, ACE inhibitors, spironolactone, and aminoglutethimide.
Epidemiology
Primary deficiency of aldosterone (hyperreninemic hypoaldosteronism) is usually caused by adrenal insufficiency and was described earlier. Secondary aldosterone deficiency caused by hyporeninemic hypoaldosteronism is also called type IV renal tubular acidosis. It most often occurs in middle-aged men, and chronic renal insufficiency is found in 80% of cases. Diabetes mellitus occurs in 50% of patients. Persons with diabetes are predisposed to hyperkalemia because of hyperglycemia and low insulin levels, which produce an extracellular flux of potassium. Autonomic neuropathy may also be a factor in the acquisition of the hyporeninemic state.
History and Physical Examination
The clinical signs of hypoaldosteronism are caused by salt wasting and hyperkalemia. The hyperkalemia can lead to muscle weakness, muscle cramps, and cardiac arrhythmias. Salt wasting can cause orthostatic blood pressure changes and hyponatremia, which can lead to mental confusion.
Diagnostic Studies
LABORATORY FINDINGS
The diagnosis of hypoaldosteronism is made by measuring the PRA and the aldosterone level in the blood during a period of salt restriction. If PRA is high, then hypoaldosteronism is an isolated defect; if PRA is low, hypoaldosteronism is secondary to the low renin level.
OTHER STUDIES
Hyperkalemia, when the potassium level is >5.5 mEq/L, can be seen on the electrocardiogram as peaked T waves. With further increases in the plasma potassium level >7 mEq/L, the P wave disappears, the ST segment is depressed, and the QRS complex widens, becoming sinusoidal. Untreated patients with severe hyperkalemia will present with ventricular fibrillation or cardiac standstill, unresponsive to electrical cardioversion, unless hypokalemia is corrected.
Treatment Options, Expected Outcomes, and Comprehensive Management
If hyperkalemia is mild, restriction of dietary potassium and avoidance of drugs that exacerbate hyperkalemia are useful. If dietary restrictions do not correct the hyperkalemia, mineralocorticoid replacement with 100 to 200 mcg/d of 9-alpha-fludrocortisol (Florinef) should be given. This therapy may promote edema from sodium retention and coexisting congestive heart failure or renal failure; hence, a balance between salt retention and fluid overload must be individualized. If Florinef cannot be tolerated, kaliuresis can be promoted using appropriate diuretics, such as chlorthalidone or hydrochlorothiazide. Sodium polystyrene sulfonate (Kayexalate), a cation exchange resin, removes potassium, but it also increases the sodium load, is expensive, and is difficult to use in the outpatient setting.
Teaching and Self-Care
Patients with hypertension often consume products advertised as being low in sodium. Nonetheless, patients with hyperkalemia or mild renal insufficiency should avoid low-sodium foods because they often contain high amounts of potassium. A healthy lifestyle, as previously described, should be encouraged.
![]() CLINICAL WARNING:
CLINICAL WARNING:
Potassium restrictions must be taught. This includes warning about specific medications, including over-the-counter drugs, as well as foods that are high in potassium. Potassium levels should be monitored.
![]() CLINICAL WARNING:
CLINICAL WARNING:
Patients with diabetes mellitus are at higher risk of becoming hyperkalemic because insulin enables the transport of glucose and potassium into cells. Patients with poorly controlled glucose levels, who are absolutely or relatively insulin-deficient, can experience cardiac arrhythmias and arrest from hyperkalemia. Therefore, after starting medications that inhibit aldosterone activity in patients with diabetes (e.g., spironolactone, ACE inhibitors, NSAIDs), potassium levels should be monitored, looking for hyperkalemia.
Hyperkalemic crisis, where a high serum level of potassium (>7 mEq/L) causes electrocardiogram changes (peak T-waves and arrhythmias), is treated emergently. Calcium gluconate (10–20 mL of a 10% solution) is given to counteract the effect of hyperkalemia on the heart. Then, glucose and insulin (25–50 g/hr glucose and 5 units of regular insulin intravenously every 15 minutes) are used to drive potassium into cells. Hemodialysis can also be used once the acute crisis is reversed.
 PHEOCHROMOCYTOMA
PHEOCHROMOCYTOMA
Catecholamine-secreting tumors of the adrenal medulla, and rarely from extra-adrenal sites, are called pheochromocytomas. Extra-adrenal pheochromocytomas are referred to as catecholamine-secreting paragangliomas. Pheochromocytoma is a rare cause of hypertension. Most cases are sporadic, but some are associated with the familial syndromes of multiple endocrine neoplasia, where pheochromocytoma is found with hyperparathyroidism and medullary thyroid cancer. Pheochromocytomas can also be associated with neurofibromatosis and Von Hippel-Lindau disease.
Anatomy, Physiology, and Pathology
The medulla of the adrenal gland is formed from cells that migrate from the fetal neural crest; thus, they are related to the cells that form the sympathetic nervous system. These cells are called neuroendocrine cells and form catecholamines from the amino acid precursor tyrosine.
Because plasma catecholamines are labile, samples must be taken fasting, resting, and supine (with the needle placed 20–30 minutes before sample taking) and processed rapidly. Urinary catecholamines are greater in magnitude and give a more integrated function over 24 hours, but use of certain drugs can give false-positive and false-negative results. Methyldopa, terbutaline, and isoproterenol will give falsely elevated catecholamine values.
Epidemiology
Pheochromocytomas arise in the adrenal gland 90% of the time. They can be bilateral in 10%, especially in those with multiple endocrine neoplasia syndrome. When the tumor is located outside the adrenal gland, it is called a paraganglioma. Pheochromocytomas are the cause in <0.2% of patients with hypertension. When patients are hypertensive and have an adrenal mass or present with hypertension, episodic headaches, palpitations, and sweating, the yield increases to about 6%. Tumors are equally common in men and women and can occur at any age, although most are found in patients 30 to 40 years old, and 50% of tumors are found on autopsy and are clinically silent during life (Young & Kaplan, 2014).
History and Physical Examination
The main sign of pheochromocytoma is either sustained hypertension or paroxysmal hypertensive crises on a background of mild hypertension, and occurs in up to 90% of symptomatic patients. The classic triad of symptoms is:
 Headache
Headache
 Sweating
Sweating
 Palpitations
Palpitations
Diagnostic Studies
LABORATORY FINDINGS
The diagnosis of pheochromocytoma is based on the finding of elevated catecholamine levels, usually measuring the urinary metabolites norepinephrine and normetanephrine in a 24-hour period. Vanillylmandelic acid (VMA) levels can be elevated but are not as specific as normetanephrine levels (65% vs. 84%; Agha, Tomlinson, Clark, Holder, & Stewart, 2006; Annane, Sébille, Charpentier et al., 2002; Eckstein et al., 2007). In recent literature, where high-performance liquid chromatography (HPLC) assays were used, collection of urinary free catecholamines was as sensitive as catecholamine metabolites such as normetanephrine, and may be more sensitive (>95%) for smaller lesions with rapid catecholamine turnover (Annane, Sébille, Charpentier et al., 2002).
The 24-hour collection of the urine is done in a jug containing acid, and creatinine should also be measured to ensure that the sample is adequate. Patients need not follow a special diet for detection of pheochromocytoma, but most antihypertensive medications should be stopped if possible. If the patient is severely hypertensive after discontinuing medication, hydralazine, minoxidil, calcium channel blockers, diuretics, or ACE inhibitors can be used. Drugs that are more apt to cause false elevations or to interfere with the collections are to be avoided, including:
 Amphetamines
Amphetamines
 Catecholamines
Catecholamines
 Clonidine withdrawal
Clonidine withdrawal
 Ethanol
Ethanol
 Methyldopa, L-dopa
Methyldopa, L-dopa
 Quinidine
Quinidine
 Theophylline
Theophylline
 Tetracycline
Tetracycline
 Metyrosine
Metyrosine
 Reserpine
Reserpine
 Guanethidine
Guanethidine
VMA collections can also be affected by monoamine oxidase inhibitors and clofibrate.
Plasma catecholamines are usually not collected as an initial screening test for two reasons: tumors can have episodic secretion that would be missed, and in some patients, the stress of testing or pain during the procedure can lead to false-positive results. Nonetheless, plasma norepinephrine levels >2,000 pg/L are highly suggestive of a pheochromocytoma during a hypertensive crisis. The catabolic effects of excessive catecholamines and decreased tissue perfusion promote lactic acidosis, and a lactate level >5 mmol/L should increase the suspicion of a pheochromocytoma (Eisenhofer et al., 2005).
Giving clonidine (0.3 mg) orally can be useful in separating patients with baseline plasma catecholamine levels above normal into those likely to have a pheochromocytoma and those with elevations caused by stress. Pheochromocytoma is suspected when, 3 hours after clonidine dosing, the plasma catecholamine levels are >500 ng/L. Provocative testing can be done with glucagon, histamine, or metoclopramide, but is not advised because these drugs can cause life-threatening cardiac responses in those with pheochromocytomas.
Special Laboratory Staining • Cells that produce catecholamines stain for neuron-specific enolase and chromogranin. In patients with pheochromocytoma, blood levels of chromogranin are often elevated.
IMAGING STUDIES
Most pheochromocytomas are found within the adrenal gland. Adrenal tumors can usually be seen by CT examination, but if there is concern that the iodine contrast material could trigger a hypertensive crisis or paroxysm, MRI also can be used. Metaiodobenzylguanidine (MIBG) is a nuclear tracer that can locate these tumors in 80% to 95% of patients, but it is not available at many sites. Isotope-labeled octreotide (Sandostatin) can also detect pheochromocytomas and is more widely available than MIBG, but it is expensive. Using MIBG or octreotide scanning, whole-body images can be obtained, so that pheochromocytomas outside the adrenal area can be detected. If nuclear scanning to locate extra-adrenal sites of pheochromocytoma is not feasible, MRI scans of the chest, abdomen, and pelvis should be done to find those rarer tumor sites (e.g., the bronchial tree, pancreatic tissue, bladder wall, or sympathetic–parasympathetic ganglia). Fludeoxyglucose-positron emission tomography (FDG-PET) is more sensitive than 1231-MIBG and CT/MRI for detection of metastatic disease.
Treatment Options, Expected Outcomes, and Comprehensive Management
Surgical resection of the pheochromocytoma reverses the manifestations of the disease, although mild hypertension may persist in 25% of cases. Laparoscopic adrenalectomy may be performed in tumors ≤6 cm in diameter and weighing <100 g. Some lesions can be multifocal or present at different times (Conzo et al., 2013).
![]() CLINICAL WARNING:
CLINICAL WARNING:
Lifelong surveillance of pheochromocytoma is necessary. It can be difficult to discern malignant from benign lesions on cytology alone. Malignancy is found in up to 13% of tumors. It is more common when the pheochromocytoma is extra-adrenal. Malignant spread to lymph tissue, liver, and bone can be seen.
Patients need preparation before surgery to avoid hypertensive crises and cardiac arrhythmias during anesthesia and hypotension on removal of the lesion. This preparation is ideally managed by an endocrine specialist. Hypertension lasting a month after surgery indicates multifocal disease or metastases or renal vasculature changes from the prior disease (discussion of surgical management is outside the bounds of this text).
Teaching and Self-Care
It may prove a diagnostic challenge to differentiate symptoms of a pheochromocytoma from those of thyrotoxicosis, drug withdrawal, anxiety disorders and panic attacks, carcinoid syndrome, paroxysmal tachycardia, mitral valve prolapse, migraine headaches, or hypoglycemia. Many patients with pheochromocytomas are asymptomatic, but those with short episodes of throbbing headache, palpitations, and sweating are more likely to have these tumors than those with a history of long-lasting headaches and incapacitation.
Clinical Pearls
 Patients with pheochromocytoma may want to lie down during an attack, whereas those with panic and anxiety disorders tend to want to leave the site of an attack.
Patients with pheochromocytoma may want to lie down during an attack, whereas those with panic and anxiety disorders tend to want to leave the site of an attack.
The body’s ability to respond to stress involves the HPA and the efferent sympathetic nervous system. Women may experience irregular or missed menstruation during stress. Stress can cause alterations in thyroid hormones, such as euthyroid sick syndrome. Thus, stress management, relaxation techniques, and regular aerobic exercise can help reduce hormonal alterations and decrease symptoms and signs associated with excessive catecholamine release in the absence of a known adrenal tumor.
![]() CLINICAL WARNING:
CLINICAL WARNING:
The hypertensive crisis caused by a pheochromocytoma leads to symptoms that could be confused with stroke, myocardial infarction, or sepsis. Crisis can be precipitated by activity (bending over, urinating, or defecating) or by exposure to certain drugs (histamine, tyramine, glucagon, naloxone, metoclopramide, ACTH, tricyclics, and phenothiazines). Crisis can also occur spontaneously or from hemorrhage within the tumor. Patients present with severe hypertension, arrhythmias, and headache. There may be anxiety and confusion with hypertensive encephalopathy. Some patients can present with myocarditis, a dilated congestive cardiomyopathy that can be accompanied by pulmonary edema. If shock intervenes from arrhythmias or sudden vasodilation from epinephrine secretion, acute bowel obstruction could result from bowel ischemia.
Community Resources
 Addison News, 6142 Territorial, Pleasant Lake, MI 49272; (517) 769–6891; www2.dmci.net/users/hoffmanrj: Education and support for persons with Addison’s disease
Addison News, 6142 Territorial, Pleasant Lake, MI 49272; (517) 769–6891; www2.dmci.net/users/hoffmanrj: Education and support for persons with Addison’s disease
 National Library of Medicine: www.nlm.nih.gov/medlineplus/healthtopics.html
National Library of Medicine: www.nlm.nih.gov/medlineplus/healthtopics.html
 Hormone Health Network: www.hormone.org
Hormone Health Network: www.hormone.org

 National Institute of Diabetes and Digestive and Kidney Diseases: www.niddk.nih.gov
National Institute of Diabetes and Digestive and Kidney Diseases: www.niddk.nih.gov
 National Cushing’s Association, 4645 Van Nuys Blvd., Sherman Oaks, CA 91403; 818-788-9239: Education and support for persons with Cushing’s disease
National Cushing’s Association, 4645 Van Nuys Blvd., Sherman Oaks, CA 91403; 818-788-9239: Education and support for persons with Cushing’s disease
 Cushing’s Support and Research Foundation, Inc., 65 East India Row, Suite 22B, Boston, MA 02110; http://world.std.com/csrf/: Information and support for persons with Cushing’s disease
Cushing’s Support and Research Foundation, Inc., 65 East India Row, Suite 22B, Boston, MA 02110; http://world.std.com/csrf/: Information and support for persons with Cushing’s disease
 Brain-Pituitary Foundation of America, 281 E. Moody Ave., Fresno, CA 93720-1524; 209-434-0610
Brain-Pituitary Foundation of America, 281 E. Moody Ave., Fresno, CA 93720-1524; 209-434-0610

Full access? Get Clinical Tree


