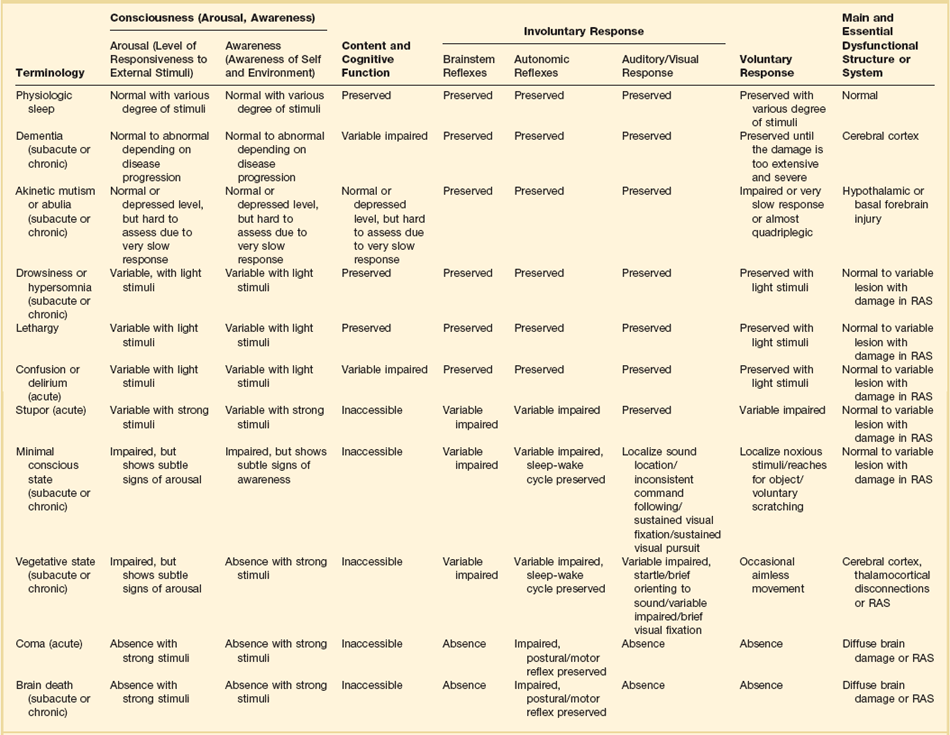
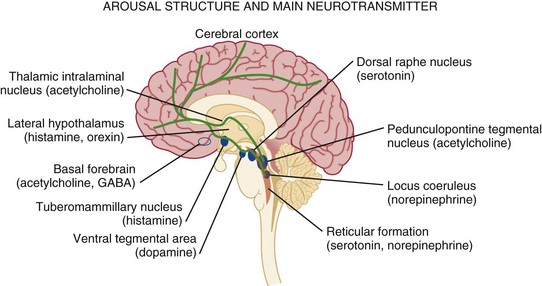
61 Alteration of consciousness is a frequent admission diagnosis to critical care services. Most patients require immediate and often extensive diagnostic workup as both time to diagnosis and treatment initiation are decisive factors for brain recovery. To the public, the portrayal of coma is overly optimistic as 89% of “comatose” patients in a meta-analysis of 64 characters in American soap dramas regained consciousness and around 92% “survived” coma.1 In contrast, medical experience does not compare as favorably, as only about 50% of patients survive in an unselected coma population.2–4 Therefore, defining coma in a succinct and operational manner will provide a deeper understanding of the depth of brain injury and avoid miscommunications and unrealistic prognostications. When evaluating a patient’s mental status, the state of consciousness should be viewed as a continuum ranging from the patient who has full alertness and cognitive lucidity to the deeply comatose patient; it is not an all-or-nothing phenomenon.5 An all-or-nothing approach limits the interpretation of remaining brain function and, hence, diagnostic certainty. Furthermore, the duration and time development of coma are helpful diagnostic features and should complement the quantitative and qualitative assessment of consciousness. Standard classification systems categorize consciousness based on systematic testing of arousal, awareness, and content. Table 61.1 classifies various abnormalities of consciousness, and the following section delineates clinically important syndromes. Coma (from Greek κ The vegetative state is characterized by the complete absence of behavioral evidence for self- or environmental awareness. This state can follow coma and identifies a state in which brainstem and diencephalic (thalamic) activity is present to a degree that clinical signs of spontaneous or stimulus-induced arousal and sleep-wake cycles are observed. Patients often show blink responses to light; intermittent eye movements (sometimes erroneously interpreted as following objects or looking at family members); stimulus-sensitive automatisms such as swallowing, bruxism, and moaning; or primitive motor responses. If this state lasts longer than 30 days, it is referred to as persistent vegetative state (PVS) and is used as a descriptive clinical syndrome rather than a disease-specific entity. The most common causes include cardiac arrest, head trauma, severe brain infections, and various causes of thalamic injury. Vegetative states can also be seen in the terminal phase of degenerative illnesses such as Alzheimer’s disease. Ambiguous terms for PVS such as apallic syndrome and neocortical death should be avoided.2,7 Minimal conscious state can be diagnosed in patients displaying some but often inconsistent behavioral evidence of awareness of the environment, but they cannot communicate their content and are unable to follow instructions reliably.8 It describes a large group of patients who are different from vegetative patients in that they demonstrate some signs of awareness of themselves and their surroundings, albeit inconsistently. Consciousness, which consists of three main parts, largely depends on the integrity of brain structure for arousal (Fig. 61.1). The ascending reticular activating system (ARAS) is the lowest order arousal system, consisting of a set of brainstem nuclei located within the brainstem and interconnected by neuronal circuits. The ARAS relays arousal signals to the more rostral thalami, which, in turn, act as hemispheric gatekeepers of arousal and regulate consciousness and sleep-wake transitions. In turn, thalamic activated cerebral cortices allow cognitive processing, which then determines the overall content of consciousness.6 Structural damage to or metabolic-chemical derangements of any or all of these elements will affect consciousness. Important components of the ARAS9–11 include the midbrain reticular formation, mesencephalic nuclei (dorsal raphe nucleus, pedunculopontine tegmental nucleus, locus coeruleus), and ventral tegmental areas. Those areas have rich connections to thalamic intralaminar nuclei and further frontally reaching projections to the tuberomammillary nucleus, lateral hypothalamus, and basal forebrain. The ascending arousal system of the brainstem contains different neurotransmitter systems. Main cholinergic projections include the ascending mesopontine tegmental and basal forebrain pathways projecting to the thalami and to virtually all subcortical and cortical structures. These projections usually function as synaptic facilitators but at times can act as depressors of transmission.12 Glutamate is the main transmitter influencing the firing patterns of tegmental cholinergic neurons.13 The adrenergic component of the ARAS is closely associated with the noradrenergic neurons of the midbrain locus coeruleus. It runs in parallel with cholinergic projections rostrally to the cortical areas but also descends caudally within the spinal cord.14 Hypocretin/orexin neurons within the hypothalamus activate both adrenergic and cholinergic pathways and coordinate activity of the entire ARAS system, enhancing complementary and synergistic control of arousal and locomotion.15 Histamine-releasing cells from the hypothalamus and tuberomammillary neurons are active during the qualitative activation needed for cognition and EEG activation, but they remain silent during sleep.16–18 The main inhibitor of the arousal system is γ-aminobutyric acid (GABA), without which normal sleep does not occur. The GABAergic circuitry is located in the midbrain and pons, but its activity is regulated and sustained by forebrain structures.19 It is thought that the GABAergic system’s main function is to contain and channel the spread of arousal, both spontaneously and in response to a stimulus. GABAergic processes are responsible for the occurrence of paradoxical sleep (deep sleep characterized by a brain wave pattern similar to that of wakefulness, rapid eye movements, and heavier breathing but without motor responses). The name “waking neurons” has been given to the serotonergic dorsal raphe nucleus in the midbrain,20 which receives convergent excitatory input from the noradrenaline, histamine, and hypocretin/orexin arousal systems21,22 as the main activator of the ARAS. Cortical signals that pass through the striatum (caudate and putamen) are refined by the action of dopamine. Dopaminergic neurons are tonically active and stimulate or inhibit cortical neurons depending on various environmental influences (pain, hunger, etc.).23,24 It is thought that many sensory signals that reach conscious awareness pass through this basal ganglia loop with dopamine as the modulating neurotransmitter. Koch and colleagues suggested that coalitions of neurons compete to dominate conscious thoughts and activities at any given time point and, hence, dopaminergic innervation may facilitate the transient domination of conscious awareness by certain sets of neuron coalitions.25 Therefore, it seems that arousal is an event with a defined time distribution, which is tightly orchestrated by a few key neurotransmitters. As we know, there is no singular “arousal neurotransmitter.”10,26 There are four major pathologies that can cause severe, acute, and global reductions of consciousness.6,27 (1) One of these pathologies is the presence of diffuse, global, or extensive multifocal bilateral dysfunction of the cerebral cortex. In this injury mechanism, the cortical gray matter is diffusely impaired and so are cortical-subcortical excitatory feedback loops. The clinical examination will reveal disinhibited autonomic brainstem reflexes, which have also been described as “reticular shock.” (2) Another is injury to the paramedian gray matter from the level of the nucleus parabrachialis of the pons (tegmentum) and reaching to the midbrain pretectal areas and ventral posterior hypothalamus. An injury of this type damages the ascending arousal system and normal cortical activation. The affected structures are predominantly the paramedian gray matter, extending rostrally from the level of the nucleus parabrachialis of the pons (tegmentum) and reaching rostrally as far as the adjacent pretectal area and ventral posterior hypothalamus. (3) The widespread disconnection of the cortex from subcortical activating mechanisms acts pathophysiologically to produce effects similar to both of the previously mentioned conditions. (4) Finally, cortical and subcortical arousal mechanisms can be affected by a variety of diffuse disorders and to various degrees—for example, metabolic encephalopathy in a patient with acute liver failure. A brief neurologic examination is mandatory before any sedative or paralytic required for intubation is administered. The key points of a rapid neurologic examination are the following:28 • Assessment of level of arousal and level of consciousness (i.e., coma, stupor, lethargy) • Pupillary size and response to light • Abnormalities of eye movements (i.e., disconjugate eyes; unilateral gaze paralysis; lack of voluntary movements) • Grimacing and motor responses to noxious stimulation The preferred route of emergency intubation is orotracheal, which can be performed rapidly, safely, and reliably with inline stabilization of the neck in patients with suspected cervical spine injury. Intubation causes intense reflexive cardiovascular stimulation that may lead to a deleterious elevation of intracranial pressure (ICP). Therefore, the patient should be adequately sedated. Etomidate is the preferred sedative in patients with suspected ICP elevation as it reliably facilitates induction in less than 1 minute with a duration of action of 4 to 6 minutes. Propofol is another anesthetic agent that does not increase ICP; however, its hypotensive effects, which can ultimately decrease cerebral perfusion pressure, often limit its use. Both medications result in a dose-dependent decrease of cerebral metabolic rate that reduces cerebral blood flow and ICP. Ketamine has a fast onset, but it may elevate ICP and should generally be avoided. Midazolam can alter the patient’s mental status and may prohibit a postintubation examination. Drug overdose is the largest single cause (30%) of coma in the emergency department. Most drug overdoses are treated by supportive measures alone. Certain antagonists, however, specifically reverse the effects of coma-producing drugs. Intravenous naloxone (0.4 to 2 mg) is used as “test” antidote for opiate-induced coma, and it acts as a µ-receptor antagonist. Caution is needed as the reversal of narcotic effect may precipitate acute withdrawal in an opiate addict. In suspected opiate coma, the minimum naloxone dose (not completely reversal) should be given to establish the diagnosis by pupillary dilation and to reverse depressed breathing and consciousness. Due to its short half-life, patients who respond to naloxone reversal may require additional doses or a continuous infusion to avoid rebound sedation and respiratory depression. Intravenous flumazenil antagonizes benzodiazepine-induced coma and can be administered in 0.2-mg doses every minute for a maximum of 1 mg. In patients who initially respond to flumazenil reversal but experience recurrent sedation, flumazenil 1 mg may be redosed every 20 minutes with a max of 3 mg/hour.29 Careful consideration should be given prior to administration of flumazenil as patients may experience benzodiazepine-withdrawal seizures. Thus, whereas naloxone is commonly used with minimal serious adverse effects, the use of flumazenil should be restricted to select, low-risk cases. The sedative effects of drugs with anticholinergic properties, particularly tricyclic antidepressants, can be reversed with 1 to 2 mg physostigmine intravenously (duration of action about 45 to 60 minutes). Pretreatment with 0.5 mg atropine will reduce the risk for symptomatic bradycardia. Of note, only full awakening is characteristic of an anticholinergic drug overdose because physostigmine has nonspecific arousal properties. Hyperthermia is detrimental in brain injury as it increases brain metabolic demands and facilitates secondary brain injury.30 Elevated temperature greater than 104° F (40° C) requires immediate, lifesaving cooling measures, even before the underlying cause is determined and treated. In 2002, two research groups independently published that lowering the body temperature to 33° C for 12 or 24 hours in comatose survivors of cardiac arrest resulted in nearly doubling the number of patients being discharged home or to rehabilitation. Generally speaking, no patient with acute brain injury should be allowed to be hyperthermic, and modern technology provides a variety types of noninvasive and invasive cooling equipment suitable to maintain any target core temperature desired. Core temperatures of less than 93° F (34° C) on admission should be slowly elevated to above 35° C to reduce the risks of unwanted side effects from uncontrolled, persistent hypothermia. A systematic, detailed examination is necessary when approaching a comatose patient (Box 61.1, Table 61.2). Important findings include evidence of trauma, acute or chronic medical illnesses, ingestion of drugs (needle marks, alcohol breath), and the presence of nuchal rigidity. Table 61.2 Correlation Between Levels of Brain Function and Clinical Signs As outlined earlier, an in-depth physical and neurologic examination followed by serial neurologic evaluations (i.e., hourly) is the most important, indispensable, and readily available method of assessing a comatose patient.31 However, in coma patients the examination remains limited in detecting changes in brain function. Nevertheless, the astute clinician will carefully evaluate for changes in findings (i.e., the appearance of new asymmetry in examination or focal deficits, progressive loss of brainstem reflexes, or loss of reflex motor responses). Finding progression of impairment is of immense clinical value as it indicates a new or worsening “intracranial emergency” demanding immediate clarification and stabilization. Skilled neurologic examination is challenging and provides a limited surveillance window of brain tissue at risk. Intracranial monitoring and, if needed, repeated head imaging have become the standard of care in neurocritical care units. Correctly characterizing disorders of consciousness continues to pose interesting clinical questions and diagnostic challenges with important ethical consequences (Box 61.2). Not only may an individual patient acutely fluctuate in his or her examination findings, but recovery may take place over prolonged time periods, necessitating constant reassessments. To address this uncertainty, standardized neurobehavioral assessments have become the best tool for categorizing coma and its transitional stages. Predicting long-term outcome has significantly improved when utilizing standardized assessments. Generally speaking, clinical and electrophysiologic markers of coma and its transitional states remain unsatisfactory. The reader is reminded that larger observational studies identified a high rate of misdiagnoses (>40%),32 especially in patients in vegetative state, when assessment is made on clinical grounds only.
Coma
Concept and Terminology of Impaired Consciousness
 µα, or “deep sleep”) is a continuous state of unresponsiveness identified by an inability to arouse to vigorous (noxious) external or internal stimuli. The degree of coma can differ; lighter stages (sometimes denoted as semicoma) can be identified by brief moaning to strong stimulation and associated observed changes in autonomic function; whereas the deepest coma examination shows an absence of any response, including brainstem reflex responses (i.e., lack of oculo- and pupillomotoric responses). Some cyclic autonomic activity such as the sleep-wake cycle and changes in motor tone may coexist. A detailed discussion of coma is provided in the classic textbook by Plum and Posner.6
µα, or “deep sleep”) is a continuous state of unresponsiveness identified by an inability to arouse to vigorous (noxious) external or internal stimuli. The degree of coma can differ; lighter stages (sometimes denoted as semicoma) can be identified by brief moaning to strong stimulation and associated observed changes in autonomic function; whereas the deepest coma examination shows an absence of any response, including brainstem reflex responses (i.e., lack of oculo- and pupillomotoric responses). Some cyclic autonomic activity such as the sleep-wake cycle and changes in motor tone may coexist. A detailed discussion of coma is provided in the classic textbook by Plum and Posner.6
Neuroanatomy, Neurotransmitter, and Pathology
Pathology Seen in Patients with Impaired Consciousness
Approach to Coma
Emergency Management
Oxygenation and Intubation
Reversal of Drug Overdose
Body Temperature
Physical Examination
Structure
Function
Clinical Sign
Cerebral cortex
Conscious behavior
Speech (including any sounds)
Purposeful movement
Spontaneous
To command
To pain
Brainstem activating and sensory pathways (reticular activating system)
Sleep-wake cycle
Eye opening
Spontaneous
To command
To path
Brainstem motor pathways
Reflex limb movements
Flexor posturing (decorticate)
Extensor posturing (decerebrate)
Midbrain CN III
Innervation of ciliary muscle and certain extraocular muscles
Pupillary reactivity
Pontomesencephalic MLF
Connects pontine gaze center with CN III nucleus
Internuclear ophthalmoplegia
Upper pons
CN V
Facial and corneal
Corneal reflex-sensory
CN VIII
Facial muscle innervation
Corneal reflex-motor response
Blink
Grimace
Lower pons
CN VIII (vestibular portion) connects by brainstem pathways with CN III, IV, VI
Reflex eye movements
Doll’s eyes
Caloric responses
Pontomedullary junction pressure
Spontaneous breathing
Maintained blood pressure
Breathing and blood pressure do not require mechanical or chemical support
Spinal cord
Primitive protective responses
Deep tendon reflexes
Babinski response
Assessment of Coma: General Aspects