Chapter 10 Clinical trials in critical care
Evidence based medicine is the conscientious, explicit, and judicious use of current best evidence in making decisions about the care of individual patients. The practice of evidence based medicine means integrating individual clinical expertise with the best available external clinical evidence from systematic research.1
RANDOMISED CLINICAL TRIALS
The result of any clinical trial may be due to three factors:
THE QUESTION TO BE ADDRESSED
Every trial should seek to answer a focused clinical question that can be clearly articulated at the outset. For example, ‘we sought to assess the influence of different volume replacement fluids on outcomes of intensive care patients’ is better expressed as the focused clinical question ‘we sought to address the hypothesis that when 4 percent albumin is compared with 0.9 percent sodium chloride (normal saline) for intravascular-fluid resuscitation in adult patients in the ICU, there is no difference in the rate of death from any cause at 28 days’.2 The focused clinical question defines the interventions to be compared, the population of patients to be studied and the primary outcome to be considered. This approach can be formalised using the PICO system. PICO stands for patient, intervention, comparison and outcome. In the example above:
Trials may be designed to answer two quite different questions about the same treatment and the design will be quite different depending on the questions to be answered. An efficacy trial seeks to determine whether a treatment will work under optimal conditions whereas an effectiveness trial seeks to determine the effects of the intervention when applied in normal clinical practice. For a detailed comparison of the features of efficacy and effectiveness trials, please see Hebert et al.3
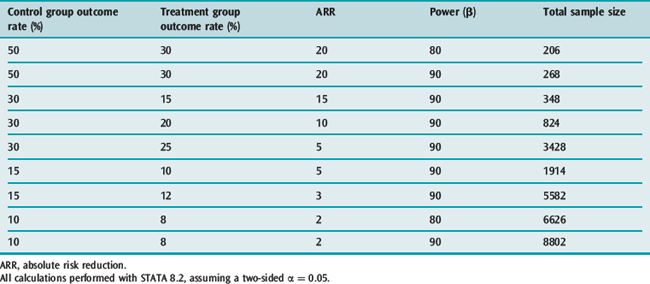
POPULATION AND SAMPLE SIZE
It is clear that many published trials addressing issues of importance in intensive care medicine are too small to detect clinically important treatment effects;4 fortunately this is now changing.2,5 This has almost certainly given rise to a significant number of false-negative results (type II errors). Type II errors result in potentially beneficial treatments being discarded. In order to avoid these errors, clinical trials have to include a surprisingly large numbers of participants. Examples of sample size calculations based on different baseline incidences, different treatment effects and different power are given in Table 10.1.
RANDOMISATION AND ALLOCATION CONCEALMENT
There are a number of benefits of using a random process to determine treatment allocation. Firstly, it eliminates the possibility of bias in treatment assignment (selection bias). In order for this to be ensured, both a truly random sequence of allocation must be produced and this sequence must not be known to the investigators prior to each participant entering the trial. Secondly, it reduces the chance that the trial results are affected by confounding. It is important that, prior to the intervention in a RCT being delivered, both groups have an equal chance of developing the outcome of interest. A clinical characteristic (such as advanced age, gender or disease severity, as measured by Acute Physiology, Age and Chronic Health Evaluation (APACHE) or Sequential Organ Failure Assessment (SOFA) scores) that is associated with the outcome is known as a confounding factor. Randomisation of a sufficient number of participants ensures that both known and unknown confounding factors (for example, genetic polymorphisms) are evenly distributed between the two treatment groups. The play of chance may result in uneven distribution of known confounding factors between the groups and this is particularly likely in trials with fewer than 200 participants.6 The third benefit of randomisation is that it allows the use of probability theory to quantify the role that chance could have played when differences are found between groups.7 Finally, randomisation with allocation concealment facilitates blinding, another important component in the minimisation of bias in clinical trials.8
Whatever method is used to produce a random allocation sequence, it is important that allocation concealment is maintained. Methods to ensure the concealment of allocation may be as simple as using sealed opaque envelopes,9 or as complex as the centralised automated telephone-based or web-based systems commonly used in large multicentre trials. Appropriate attention to this aspect of a clinical trial is essential as trials with poor allocation concealment produce estimates of treatment effects that may be exaggerated by up to 40%.10
THE INTERVENTIONS
The intervention being evaluated in any clinical trial should be described in sufficient detail that clinicians could implement the therapy if they so desired, or researchers could replicate the study to confirm the results. This may be a simple task if the intervention is a single drug given once at the beginning of an illness, or may be complex if the intervention being tested is the introduction of a process of care, such as the introduction of a medical emergency team.11 There are two additional areas with regard to the interventions delivered in clinical trials that require some thought by those conducting the trial and by clinicians evaluating the results, namely blinding and the control of concomitant interventions.
BLINDING
Blinding, also known as masking, is the practice of keeping trial participants (and, in the case of critically ill patients, their relatives or other legal surrogate decision-makers), care-givers, data collectors, those adjudicating outcomes and sometimes those analysing the data and writing the study reports unaware of which treatment is being given to individual participants. Blinding serves to reduce bias by preventing clinicians from consciously or unconsciously treating patients differently on the basis of their treatment assignment within the trial. It prevents data collectors from introducing bias when recording parameters that require a subjective assessment, for example pain scores and sedation scores or the Glasgow Coma Score. Although many ICU trials cannot be blinded, for example, trials of intensive insulin therapy cannot blind treating staff who are responsible for monitoring blood glucose and adjusting insulin infusion rates, the successful blinding of the Saline versus Albumin Fluid Evaluation (SAFE) trial demonstrated the possibility of blinding even large complex trials if investigators are sufficiently committed and innovative.2 Blinded outcome assessment is also necessary when the chosen outcome measure requires a subjective judgement. In such cases the outcome measure is said to be subject to the potential for ascertainment bias. For example, a blinded outcome assessment committee should adjudicate the diagnosis of ventilator-associated pneumonia (VAP) and blinded assessors should be used when assessing functional neurological recovery using the extended Glasgow Outcome Scale; both the diagnosis of VAP and assessment of the Glasgow Outcome Scale require a degree of subjective assessment and are therefore said to be prone to ascertainment bias.
It has been traditional to describe trials as single-blinded, double-blinded or even triple-blinded. However these terms can be interpreted by clinicians to mean different things, and the terminology may be confusing.12 We recommend that reports of RCTs include a description of who was blinded and how this was achieved, rather than a simple statement that the trial was ‘single-blind’ or ‘double-blind’.13 Blinding is an important safeguard against bias in RCTs, and although not thought to be as essential as maintenance of allocation concealment, empirical studies have shown that unblinded studies may produce results that are biased by as much as 17%.10
CONCOMITANT TREATMENTS
Concomitant treatments are all treatments that are administered to patients during the course of a trial other than the study treatment. With the exception of the study treatment, patients assigned to the different treatment groups should be treated equally. When one group is treated in a way that is dependent on the treatment assignment, but not directly related to the treatment, there is the possibility that this third factor will influence the outcome. An example might be a trial of pulmonary artery catheters (PACs), compared to management without a PAC. If the group assigned to receive management based on the data from a PAC received an additional daily chest X-ray to confirm the position of the PAC, they could conceivably have other important complications noted earlier, such as pneumonia, pulmonary oedema or pneumothoraces, and this may affect outcome in a fashion unrelated to the data available from the PAC. Maintaining balance in concomitant treatments is facilitated by blinding. When trials cannot be blinded, use of concomitant treatments that may alter outcome should be recorded and reported, so that the potential impact of different concomitant treatments can be assessed.
OUTCOME MEASUREMENT
A clinically meaningful outcome is a measure of how patients feel, function or survive.14 Clinically meaningful outcomes are the most credible end-points for clinical trials that seek to change clinical practice. Phase III trials should always use clinically meaningful outcomes as the primary outcome. Examples of clinically meaningful outcomes include mortality and measures of health-related quality of life. In contrast, a surrogate outcome is a substitute for a clinically meaningful outcome; a reasonable surrogate outcome would be expected to predict clinical benefits based upon epidemiologic, therapeutic, pathophysiologic or other scientific evidence.14 Examples of surrogate end-points would include cytokine levels in sepsis trials, changes in oxygenation in ventilation trials or blood pressure and urine output in a fluid resuscitation trial.
Unless a surrogate outcome has been validated, it is unwise to rely on changes in surrogate outcomes to guide clinical practice. For example, it seemed intuitively sensible that after myocardial infarction the suppression of ventricular premature beats (a surrogate outcome) which were known to be linked to mortality (the clinically meaningful outcome) would be beneficial. Unfortunately the Cardiac Arrhythmia Suppression Trial (CAST) trial found increased mortality in participants assigned to receive antiarrhythmic therapy.15 The process for determining whether a surrogate outcome is a reliable indicator of clinically meaningful outcomes has been described.16





Full access? Get Clinical Tree


