Protein
Gene
Human chromosome
Distribution
Tetrodotoxin sensitive
Nav1.1
SCN1A
2q24.3
CNS; heart
+
Nav1.2
SCN2A
2q24.3
CNS
+
Nav1.3
SCN3A
2q24.3
Foetal DRG
+
Nav1.4
SCN4A
17q23.3
Muscle
+
Nav1.5
SCN5A
3p22.2
Heart
−
Nav1.6
SCN8A
12q13.13
DRG; CNS
+
Nav1.7
SCN9A
2q24.3
DRG; SCG
+
Nav1.8
SCN10A
3p22.2
DRG
−
Nav1.9
SCN11A
3p22.2
DRG
−
Nax
SCN7A
2q24.3
Lung nerve
+
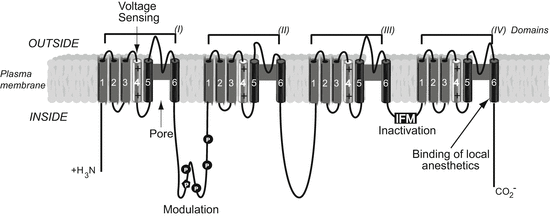
Fig. 1
Primary structure of the alpha subunit of a generic voltage-gated sodium channel within the plasma membrane lipid bilayer (in grey). Alpha helical transmembrane segments are shown as cylinders; bold lines represent polypeptide chains. P denotes sites of demonstrated protein phosphorylation by PKA (circles) and PKC (pentagon). Residues in the inner cavity of the channel pore involving the S6 segment of domains I, III and IV form the binding site for some local anaesthetic, antiepileptic and antiarrhythmic drugs, such as lidocaine, mexiletine and carbamazepine (Ragsdale et al. 1994, 1996)
2 Sodium Channels and Pain: Insights from Rodent Studies
Detection of noxious stimuli and signalling of tissue damage are physiological processes that promote animal survival. Peripheral sensory neurons, with cell bodies located within the dorsal root ganglia (DRG), express receptors that are sensitive to noxious thermal, chemical or mechanical stimuli and transduce these damaging stimuli into electrical activity through the activation of VGSCs. Within DRG, several VGSCs can be detected with differing cellular expression profiles and biophysical properties, where they work to fine-tune the signalling of noxious information to the central nervous system. Nav1.7, Nav1.8 and Nav1.9 are preferentially expressed within DRG neurons, whereas Nav1.3 expression is upregulated following nervous system injuries. Through detailed studies of Nav transgenic mice, the contribution of each channel to pain signalling is being elucidated (Table 2).
Table 2
A summary of acute, inflammatory and neuropathic pain behaviour in Nav X transgenic mice (X denotes Cre line used to tissue-specifically ablate Nav expression)
Transgenic mouse | Acute pain | Inflammatory pain | Neuropathic pain | |||
|---|---|---|---|---|---|---|
No change | Reduced | No change | Reduced | No change | Reduced | |
Nav1.7 flox × Nav1.8 Cre | Hot platea,b | Randall-Selittoa,b | Foot licking/bitinga formalin | |||
Cold plateb | Foot licking/biting acetone b | Hargreavesa CFA, NGF, Carrageenan | ||||
Hargreavesb | Hargreavesa | Von Freya CFA | ||||
Von Freya,b | ||||||
Nav1.7 flox × Advillin Cre | Hot plateb | Randall-Selittob | Von Freyb SNT | |||
Von Freyb | Foot licking/biting acetone b | |||||
Cold plateb | Hargreavesb | |||||
Nav1.7 flox × Wnt1 Cre | Cold plateb | Randall-Selittob | Von Freyb SNT | |||
Foot licking/biting acetone b | ||||||
Von Freyb | Hargreavesb | |||||
Hot plateb | ||||||
Nav1.8 KO | Hargreavesc,d | Hargreavese | Hargreavesd Carrageenan | Hargreavese Carrageenan | Hargreavesc after SNT | |
Hot platec,d,e | Randall-Selittoc,e | Von Freyd CFA | Hargreavesc,d CFA | Von Freyc,d after SNT, SNI,CCI | ||
Floxed stop DTA × Nav1.8 Crec | Von Freyc,e | Cold plateb,c,f | Foot licking and biting/liftingd formalin | Von Freyc FCA | Weight-bearingd after SNI, CCI | |
Warm waterd | Foot licking and biting/liftingc formalin, FCA | Flinching/licking/liftingd acetone after CCI, SNI | ||||
Foot licking and biting/lifting after acetone b | ||||||
Nav1.9 KO | Von Freyg,h | Von Freyd,h CFA | Hargreavesg Carrageenan, CFA, PGE2 | Von Freyd,g,h after SNI, CCI | Foot licking/flinchingd acetone after CCI | |
Hargreavesd,g | Foot licking/flinchingd,g,h,i formalin, bradykinin, αβmet-ATP, UTP | |||||
Hot plated,g,h | Hargreavesd CFA, Carrageenan | Von Freyh,i bradykinin, PGE2, IL-1β, NGF, Carrageenan | Foot licking/flinchingd,h acetone after SNI | |||
Cold plateh | Hot plateh bradykinin, PGE2, IL-1β, NGF, CFA | |||||
Warm waterd | Warm wateri Carrageenan, CFA | Weight-bearingd after SNI, CCI | ||||
Dynamic weight-bearingi Carrageenan, CFA | ||||||
Nav1.3 KO | Hargreavesj | Foot licking/bitingj formalin | Von Frey after spinal nerve ligationj | |||
Hot platej | ||||||
Von Freyj | Hargreavesj CFA | |||||
Randall-Selittoj | Von Freyj CFA | |||||
2.1 Nav1.7
Nav1.7 has seen special interest in recent years as several inherited human pain disorders, including congenital pain insensitivity, have been shown to result from mutations in this channel (see Sect. 3). The Nav1.7 channel is characterised biophysically as a tetrodotoxin-sensitive, fast-activating and a fast-inactivating channel that recovers slowly from fast inactivation (Klugbauer et al. 1995). Nav1.7 also has slow closed-state inactivation properties, allowing the channel to generate a ramp current in response to small, slow depolarisations (Cummins et al. 1998; Herzog et al. 2003). Nav1.7-positive neurons are therefore able to amplify slowly developing subthreshold depolarising inputs, such as generator potentials arising in peripheral terminals of nociceptors. Nav1.7 is expressed within DRG and trigeminal ganglia peripheral sensory neurons, as well as sympathetic neurons and olfactory epithelia (Toledo-Aral et al. 1997; Weiss et al. 2011). Nav1.7 shows particularly high expression within the soma of small-diameter DRG neurons and along the peripherally and centrally directed C fibres of these cells (Black et al. 2012). Expression can also be detected within the peripheral terminals of DRG neurons in the skin and also in the preterminal central branches and terminals in the dorsal horn, as well as at nodes of Ranvier in a subpopulation of small-diameter myelinated fibres.
In 2004, conditional deletion of Nav1.7 within Nav1.8-positive nociceptors highlighted the importance of this channel to pain behaviour, with knockout animals losing acute noxious mechanosensation and inflammatory pain (Nassar et al. 2004). In 2012, an Advillin Cre line was used to ablate Nav1.7 expression in all DRG sensory neurons, with knockout mice showing an additional loss of noxious thermosensation (Minett et al. 2012). This suggests that Nav1.7 expressed within Nav1.8-positive sensory neurons is important for acute noxious mechanosensation, whilst Nav1.7 expressed within Nav1.8-negative DRG neurons is essential for acute noxious thermosensation. The contribution of Nav1.7 to the development of neuropathic pain behaviour has also been assessed using different Cre mice to delete Nav1.7 in subgroups of cell populations. Neuropathic pain behaviour develops normally in mice where Nav1.7 has been deleted in Nav1.8-positive sensory neurons and in Advillin-positive DRG neurons (Nassar et al. 2005; Minett et al. 2012). In contrast, mice in which Nav1.7 is deleted from all sensory neurons as well as sympathetic neurons (using a Wnt1-Cre) show a dramatic reduction in mechanical hypersensitivity following a surgical model of neuropathic pain, demonstrating an important role for Nav1.7 in sympathetic neurons in the development of neuropathic pain (Minett et al. 2012).
2.2 Nav1.8
The TTX-resistant Nav1.8 sodium channel is expressed in the majority of small-diameter unmyelinated nociceptive DRG neurons (Akopian et al. 1996). Nav1.8 displays ~10-fold slower kinetics with a depolarised voltage dependence of activation and inactivation than the fast and rapidly inactivating TTX-S DRG channels (Akopian et al. 1996; Cummins and Waxman 1997). Analysis of Nav1.8 null mice shows that Nav1.8 carries the majority of the current underlying the upstroke of the action potential in nociceptive neurons (Akopian et al. 1999; Renganathan et al. 2001). Furthermore, Nav1.8 is cold resistant, meaning that it is essential in maintaining the excitability of nociceptors at low temperatures (Zimmermann et al. 2007).
In contrast to Nav1.7 global knockout mice, Nav1.8 null mice are viable, fertile and apparently normal (Akopian et al. 1999). However, pain behaviour analyses show that Nav1.8 null mice have reduced sensitivity to noxious mechanical stimuli (tail pressure), noxious thermal stimuli (radiant heat) and are insensitive to noxious cold stimuli (Akopian et al. 1999; Zimmermann et al. 2007) (Table 2). Furthermore, Nav1.8 is important in nerve growth factor-induced thermal hyperalgesia, although neuropathic pain behaviour is normal in these mice following peripheral nerve injury (Kerr et al. 2001). Transgenic mice in which Nav1.8-expressing neurons are ablated by the targeted expression of diphtheria toxin A are resistant to noxious cold and noxious mechanical stimuli but show a normal hot plate response (Abrahamsen et al. 2008). Furthermore, mechanical and thermal hyperalgesia induced by inflammatory insults is attenuated in these mice, although neuropathic pain behaviour is normal.
2.3 Nav1.9
Nav1.9 was cloned in 1998 and has a similar expression pattern within DRG to Nav1.8 (Dib-Hajj et al. 1998; Fang et al. 2002). Nav1.9 generates extremely slow persistent TTX-resistant currents and can be activated at potentials close to the resting membrane potential (Cummins et al. 1999; Dib-Hajj et al. 2002). The activation kinetics are too slow to contribute to the upstroke of an action potential, although Nav1.9 may amplify subthreshold depolarisations and lower the threshold for action potential induction (Cummins et al. 1999; Herzog et al. 2001; Baker et al. 2003). Mice lacking the Nav1.9 channel have normal sensitivity to acute mechanical and thermal noxious stimuli suggesting that Nav1.9 is not essential for setting basal pain thresholds; however, under inflammatory conditions, the channel plays a predominant role in mechanical and thermal hyperalgesia (Table 2) (Priest et al. 2005; Amaya et al. 2006; Lolignier et al. 2011).
2.4 Nav1.3
Nav1.3, like Nav1.7, is a TTX-sensitive channel with fast kinetics and a rapid recovery from fast inactivation. Unlike Nav1.7, Nav1.3 has very low expression levels in the DRG in adults, instead having a predominant expression pattern in the central and peripheral nervous system during embryogenesis (Beckh et al. 1989; Waxman et al. 1994). In humans, Nav1.3 is widely expressed in adult brain and is thought to play a role in propagating synaptic signals from dendrites to the soma and integration of electrical signals within the soma prior to the initiation of axonal action potentials (Whitaker et al. 2001). Nav1.3 levels increase in peripheral sensory neurons following nerve injury or inflammation, suggesting that this channel could play a role in pain (Waxman et al. 1994; Dib-Hajj et al. 1999). However, Nav1.3 global and nociceptor-specific knockout mice show normal neuropathic pain behaviour (Table 2) (Nassar et al. 2006).
3 Human Heritable Sodium Channelopathies
Genetic analyses of sporadic patients and families with rare inherited pain disorders have given important insights into the role of VGSCs in human pain pathways (Goldberg et al. 2012a). In recent years, the mutations that underlie several human pain disorders have been identified, and with the introduction of exome and whole genome sequencing, the pace of disease gene identification has significantly increased and will continue to do so (Table 3). With the obvious importance of the Nav family in neuronal signalling, it is perhaps unsurprising that several human disorders, including epilepsy (Nav1.1, Nav1.2 and Nav1.3), hyperkalaemic periodic paralysis (Nav1.4), Brugada syndrome (Nav1.5) and learning disability with cerebellar ataxia (Nav1.6), are caused by mutations in these channels. Likewise, inherited mutations in three of the sodium channel genes expressed in damage-sensing neurons (Nav1.7, Nav1.8 and Nav1.9) give rise to distinct human disorders with phenotypes ranging from lacerating chronic pain to complete pain insensitivity. By understanding how the gene mutations specifically affect sodium channel function, we can learn more about the aetiology of each pain disorder and also flag potential new human-validated analgesic drug targets.
Table 3
Human pain-related channelopathies
Protein | Gene | Loss of function phenotype | Gain-of-function phenotype |
|---|---|---|---|
Nav1.7 | SCN9A | Channelopathy-associated insensitivity to pain (2006) | Primary erythromelalgia (2004) |
Hereditary sensory and autonomic neuropathy type IID (2013) | Paroxysmal extreme pain disorder (2006) | ||
Painful small-fibre neuropathy (2011) | |||
Nav1.8 | SCN10A | None reported in humans | Painful small-fibre neuropathy (2012) |
Nav1.9
Related posts:
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|



