Chapter 45 Cerebral protection
The cerebral circulation is arguably the most important and most vulnerable in the body. Arrest of the circulation for only a few minutes can cause neuronal death. The concept of cerebral protection has been engaged across a broad spectrum of clinical scenarios. It has been incorporated into the prophylaxis, treatment and subsequent management of ischaemia and infarction and even into attempts to ameliorate postischaemic or anoxic damage following cardiorespiratory resuscitation. A complete review is beyond the scope of this chapter, but an understanding of the current approaches to cerebral protection is certainly helpful in the management of cerebral insults.
NORMAL BRAIN PHYSIOLOGY
COLLATERAL BLOOD SUPPLY
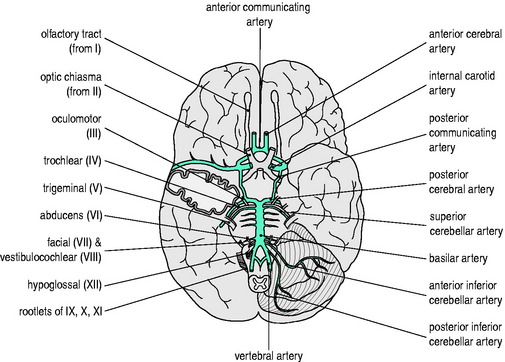
An elaborate vascular architecture is designed to ensure adequate CBF. The anterior cerebral circulation is provided by bilateral internal carotid arteries as they each divide into anterior and middle cerebral arteries. They provide approximately 70% of the cerebral circulation and supply the anterior cerebrum (frontal, parietal and temporal lobes) and anterior diencephalon (basal ganglia and hypothalamus). The remaining 30% of the cerebral circulation is provided posteriorly by the vertebrobasilar system which runs the length of the posterior fossa, supplying the brainstem, cerebellum and posterior portion of the cerebrum (occipital lobes) and diencephalon (thalamus). These two circulations are joined by communicating arteries, ultimately forming the circle of Willis (Figure 45.1). This joining of the anterior and posterior circulations at the base of the brain is the major component of cerebral vasculature in humans. Between these arterial distributions are watershed zones fed by leptomeningeal connections. In addition, persistent fetal arteries can infrequently provide collateral routes between the anterior and posterior arterial systems in the brain.
CEREBRAL BLOOD FLOW
CEREBRAL PERFUSION PRESSURE
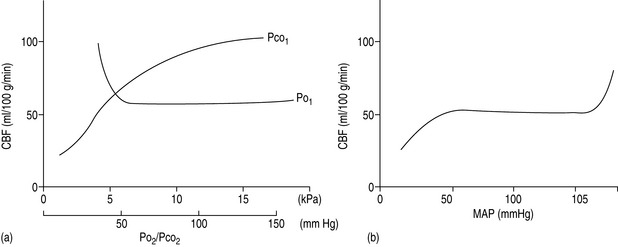
The cerebral vessels change diameter inversely with changing perfusion pressure: as CPP rises, the vessels constrict and as CPP falls the vessels dilate, such that blood flow is kept constant over a wide range of CPP (Figure 45.2a). This pressure autoregulation is thought to be controlled by local myogenic responses of the vessel wall to changes in intra-arterial pressure. At pressures above and below this range of 6.7–20 kPa (50–150 mmHg), cerebral perfusion becomes pressure-passive and increases or decreases in direct proportion to changes in CPP. The autoregulatory range varies with age, being shifted to the left in newborns and to the right in those with chronic hypertension. The latter is important to remember to avoid overtreating systolic blood pressure in such patients and thus incur the risk of cerebral ischaemia at the lower limits of autoregulation. Alternatively, cerebral perfusion above normal can be caused by acute hypertension overcoming the upper limits of autoregulation. This may lead to cerebral oedema secondary to increased hydrostatic pressures (hypertensive encephalopathy) and potentially lead to seizures or cerebral haemorrhage.
PaO2 AND PaCO2 EFFECTS
A second group of factors control CBF through an influence on the local metabolic milieu. Prominent in this mechanism are oxygen and carbon dioxide. Arterial content or partial pressure of oxygen in the normal or hyperoxic ranges causes very little change in CBF. Perhaps this represents a demand for another nutrient (i.e. glucose) or a need to remove waste products (i.e. carbon dioxide or metabolic acid). With the onset of hypoxaemia (PaO2 60 mmHg or 8 kPa), there is a prompt increase in CBF proportional to the decrease in blood oxygen content, in order to maintain oxygen delivery constant (Figure 45.2b).
There is also a direct relationship between CBF and PaCO2, such that cerebral perfusion increases with increasing PaCO2 (Figure 45.2b). This probably represents the need of the brain to maintain homeostatic pH by removing metabolic breakdown products more efficiently by increased blood flow. Unlike the response to oxygen, the CBF response to changes in PaCO2 is dramatic in the physiological range, such that for every 0.13 kPa (1.0 mmHg) change in PaCO2 there is a 1–2 ml/min per 100 g tissue change in CBF. Therefore, an increase in PaCO2 to 10.6 kPa (80 mmHg) will increase CBF to approximately 100 ml/min per 100 g and a decrease in PaCO2 to 2.7 kPa (20 mmHg) will decrease CBF to 25 ml/min per 100 g. Thus:
GLOBAL HYPOXIC/ISCHAEMIC INSULTS
These types of insult are caused by hypoxaemia and cardiovascular insufficiency or arrest, respectively. These are usually sudden, short and severe. If there is to be recovery, prompt return of oxygen delivery and spontaneous circulation are necessary. The recovery may be variable, depending on the severity and duration of the insult and the selective vulnerability of the cell types involved. Different mechanisms are responsible for reversible loss of cellular function and for irreversible cell death and there are also differences between the mechanisms that cause death of neurones, glia and endothelial cells. After 4–6 minutes of complete global ischaemia, there are signs of permanent histological damage in selective neuronal populations and the beginnings of neurological deficits in survivors. Outcome worsens significantly after 15 minutes of global ischaemia.1
BRAIN ISCHAEMIC AND INFARCTION PROCESSES
Changes in normal physiology begin to occur when blood flow is reduced.
EFFECTS OF ISCHAEMIA
This begins a cascade of events resulting in eventual cell death:
Other effects within the cell influence DNA and RNA production, hence inhibiting protein production. This may explain why cellular and clinical recovery is partial, even with restoration of ionic equilibrium and near-normal ATP levels after successful reperfusion. Necrosis is thought to occur in the core of the cerebral infarct following acute vascular occlusion, with further neurodegeneration occurring more slowly in the penumbra, by apoptosis or release of various immunological mediators.2
EXTRACELLULAR EFFECTS
Leukocytes are thought to be major contributors to reperfusion injury in that:
MANAGEMENT
In cases of head trauma, it is very important to prevent secondary brain injury. Reviews of intensive care practice have resulted in recommendations for treatment in this group of patients.3 These involve:
HYPERTENSION
Most patients have elevated blood pressure in the early phase after an acute ischaemic stroke. The mechanism and effects of this blood pressure elevation are not well understood and the benefits of intervention remain debated. Most experts would not advocate antihypertensive treatment in the acute phase after stroke, unless the blood pressure is particularly high. Currently hypertension (systolic blood pressure > 180 mmHg) should be treated if thrombolytics are to be administered, based on the trials involving these agents in which the blood pressure was lowered prior to treatment.4 Ongoing studies such as Blood Pressure in Acute Stroke Collaboration (BASC) should help define optimal management.
HYPERGLYCAEMIA
Hyperglycaemia has also been associated with an increased mortality and reduced functional outcome after stroke irrespective of whether the patient has a diabetic history or not.5 Proposed explanations for this are:
Hyperglycaemia may result in a hypercoaguable state, with decreased plasma fibrinolytic activity, which may delay reperfusion and increase haemorrhagic infarct conversion in tissue plasminogen activator-treated patients.6
Insulin, as well as having a glucose-lowering effect, has also been shown to be directly neuroprotective and this is the subject of a number of ongoing trials7 (Treatment of Hyperglycaemia in Ischaemic Stroke (THIS) trial, the Glucose Regulation in Acute Stroke Patients (GRASP) trial and the Glucose Insulin Stroke Trial UK (GIST-UK).
HYPERTHERMIA
An increased temperature increases cerebral metabolism, oxygen requirements, CBF and ICP. Hyperpyrexia following acute stroke adversely influences stroke severity, infarct size, functional outcome and mortality.8 A raised temperature should, therefore, be treated aggressively and any evidence of infection identified early and treated with appropriate antibiotics.



Full access? Get Clinical Tree


