CHAPTER 4 Cardiovascular Physiology
Fetal circulation
Anatomy
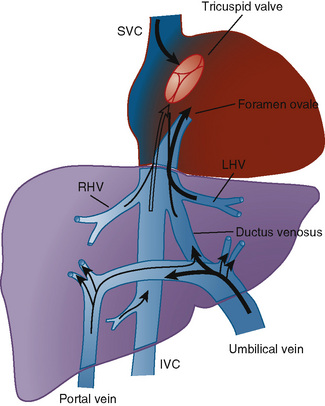
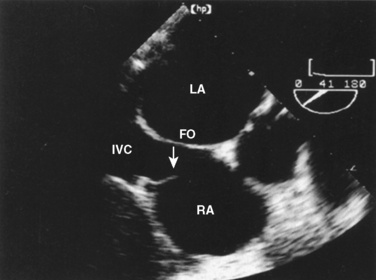
The organ of prenatal respiration is the placenta. It is a large, low-resistance circuit that has an enormous influence on the pattern of fetal blood flow. The lungs are almost completely excluded from fetal circulation, and three special shunts (the ductus venosus, the foramen ovale, and the ductus arteriosus) allow the most oxygenated blood to perfuse the heart and brain (Fig. 4-1). Two umbilical arteries originate from the internal iliac arteries and deliver fetal blood to the placenta. One umbilical vein carries oxygenated blood from the placenta to the fetus. When umbilical venous blood approaches the liver, it can take two pathways. It is estimated that 50% to 60% of umbilical venous blood bypasses the liver via the ductus venosus, while the remainder perfuses the left lobe of the liver. Blood flow to the right lobe of the liver is predominantly from the portal circulation. The right and left hepatic veins, along with the ductus venosus, merge into the suprahepatic inferior vena cava (IVC) (Fig. 4-2). Bypassing the high-resistance hepatic microcirculation, umbilical venous blood in the ductus venosus remains not only more oxygenated, but it also flows at a higher velocity. Within the suprahepatic IVC there are now two streams of blood. The stream of blood with higher velocity is derived from the ductus venosus and drainage from the left hepatic vein. The stream of blood with slower velocity consists of drainage from the right hepatic vein mixed with venous return from the abdominal IVC. Upon entering the right atrium, these two streams of blood diverge. The blood with higher velocity is primarily directed across the foramen ovale to the left atrium. This right-to-left shunt is possible because left atrial pressure is low due to minimal pulmonary venous return. The Eustachian valve is a flap of tissue at the junction of the IVC and the right atrium. It functions to help direct the higher-velocity stream of blood across the foramen ovale and into the left atrium (Fig. 4-3). The lower-velocity stream of blood crosses the tricuspid valve and is ejected by the right ventricle. This anatomic arrangement allows the most oxygenated blood from the umbilical vein to bypass the liver and the right side of heart.
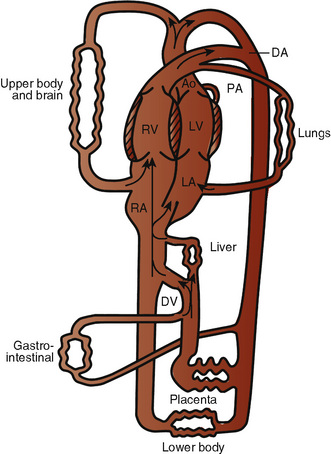
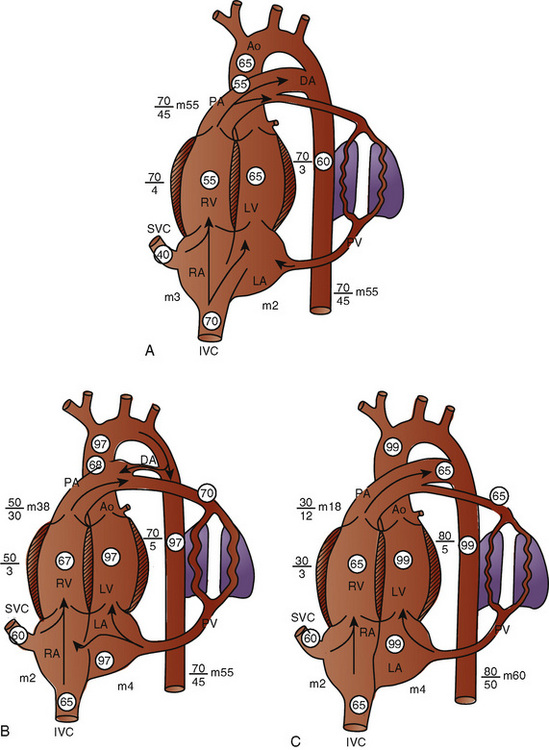
The key features of fetal circulation are shown in Figure 4-4 and listed below:
Fetal Cardiac Output
In the fetus, right and left ventricular outputs are not equal. The ventricles, acting in parallel, pump different amounts of blood, and organs receive blood flow from both ventricles. Thus, it is customary to refer to the combined ventricular output (CVO) of fetal circulation. The fetal CVO is estimated at 400 mL/kg per minute, a value that is similar to that of the neonate but almost threefold higher than it is in adults. The ratio of right ventricular to left ventricular output is approximately 1.3:1 (Kenny et al., 1986). The volume load borne by the right ventricle combined with the high fetal PVR results in significant hypertrophy. After birth, the right ventricle remodels over time under the influence of a series circulation and lowered PVR (Fig. 4-5). The demands of a parallel circulation result in increased myocardial work superimposed on the demands of a fetus having to grow and develop in a cyanotic milieu. Nevertheless, the fetal circulation is an efficient arrangement with adequate physiologic reserve. This is demonstrated by the full spectrum of congenital heart lesions that are well tolerated in utero.
Oxygen Delivery
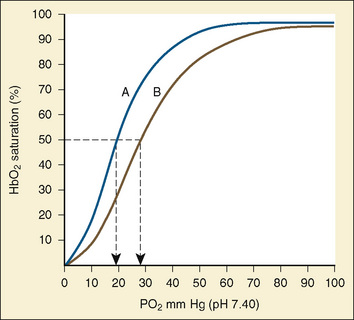
Oxygen transport must be achieved in a relatively hypoxic environment (Lister et al., 1979). How does the fetus ensure adequate oxygen delivery? Fetal hemoglobin (HbF) has unique properties that allow the fetus to transport oxygen despite a low Po2. Approximately 80% of fetal hemoglobin is HbF compared with an adult who has over 90% adult hemoglobin (HbA). The Pao2 at which Hb is 50% saturated is called the P50. Fetal hemoglobin (HbF P50:19 mm Hg) is shifted to the left in comparison with adult hemoglobin (HbA P50:26 mm Hg). A low level of 2,3-diphosphoglycerate (2,3-DPG) and the decreased affinity of HbF for 2,3-DPG cause the leftward shift of HbF. In vitro, HbF has a sigmoidal oxygen dissociation curve similar to that of HbA. The result is that for any given Po2, the oxygen saturation is higher for HbF than for HbA.
Assuming a Pao2 of 30 mm Hg in the blood ejected by the left ventricle, this value falls in the steepest part of the oxygen-hemoglobin dissociation curve. At a Pao2 of 30 mm Hg, fetal hemoglobin would be approximately 70% saturated, and adult hemoglobin would only be 50% saturated (Fig. 4-6). Using the equation for oxygen content of the blood (Cao2) demonstrates how the fetus can achieve levels of oxygen transport that are near those of an adult. Oxygen content of blood is defined by the following equation, Sao2 is the arterial oxygen saturation of hemoglobin, and Hb is the hemoglobin concentration in g/dL:
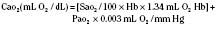
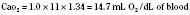
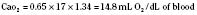
Transitional circulation
At birth, the fetus must make a transition to an adult circulatory system. The fact that the vast majority of newborns make this transition smoothly does not mean that the changes required are inconsequential. On the contrary, the events precipitating the transition from fetal to adult circulation are profound and immediate, requiring the fetus to make dramatic changes to ensure survival. The primary events that occur at birth are the clamping of the umbilical cord and initiation of breathing, with inflation of the lungs with air. These changes markedly alter the resistances in the cardiovascular system, changing the pattern of blood flow through the three vital shunts that characterize the fetal circulation. The fetus moves from a circulation that functions in parallel to one that is in a series. The lungs must now become the organs of oxygen supply and ventilation. Lung inflation and increased oxygen tension lower PVR dramatically, causing increased pulmonary blood flow and increased blood return to the left atrium. Cord clamping removes the low-resistance placenta from the circulation system and raises SVR. Left atrial pressure now exceeds right atrial pressure, closing the flap of tissue covering the foramen ovale. Normal intracardiac pressures keep the tissue flap over the foramen ovale closed, and over weeks it will completely seal. However, permanent occlusion does not occur in up to 25% of adults who retain a small defect or probe patency of the patent foramen ovale (PFO) (Hagen et al., 1984). Left atrial pressure rises for two reasons. First, left atrial volume increases significantly because of the increase in pulmonary blood flow. Second, left-sided pressures in the heart rise because of the increase in SVR. With the increase in systemic blood pressure and fall in PVR, flow through the ductus arteriosus becomes initially bidirectional and then very quickly evolves into a left-to-right shunt. Within the first hours of life the ductus arteriosus begins to close under the influence of the increased oxygen tension, loss of placental prostaglandins, and the more alkalotic environment of the newborn. Permanent closure of the ductus arteriosus takes weeks to occur.
Cardiac output in utero is considered to be the combined output of both ventricles and has been estimated at 400 mL/kg per minute. The right ventricle does more of this work, resulting in its hypertrophied state at birth. After birth, right ventricular work decreases because the volume load is reduced and PVR falls. In a series circulation, the cardiac output is the equal volume of blood ejected by each ventricle. Newborn cardiac output is the same as that in utero but by convention is calculated as 200 mL/kg per minute, which is the output of each ventricle (Lister et al., 1979). In the transition to postnatal life, it is the left ventricle that must cope with increased demands. Left ventricular output increases two- to threefold. This is accomplished by an increase in the left ventricular preload and stroke volume and heart rate. The volume load increase and rise in SVR represent a significant increase from the fetal state (Anderson, 1996). Additionally, as a result of the high beta stimulation associated with labor and delivery, heart rate increases and is near maximum. The newborn must maintain a high cardiac output because its metabolic rate (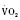 :6-7 mL O2/kg per minute) is double that of an adult (Anderson, 1990). With a large surface-to-mass ratio, the newborn is at a significant disadvantage for maintaining temperature. Its compensation is to use its high metabolic rate to generate heat for temperature homeostasis (Hill and Rahimtulla, 1965). The remainder of the increased
:6-7 mL O2/kg per minute) is double that of an adult (Anderson, 1990). With a large surface-to-mass ratio, the newborn is at a significant disadvantage for maintaining temperature. Its compensation is to use its high metabolic rate to generate heat for temperature homeostasis (Hill and Rahimtulla, 1965). The remainder of the increased 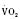 is devoted to growth and the oxygen requirement of the brain, which is proportionally much larger in newborns.
is devoted to growth and the oxygen requirement of the brain, which is proportionally much larger in newborns.
Persistence of fetal circulation most commonly occurs in instances of severe prematurity with respiratory distress syndrome (RDS). Hypoxemia is a potent stimulus, maintaining a patent ductus arteriosus (PDA). Flow through the ductus arteriosus remains possible as long as it is patent. The direction of that flow depends solely on the relative resistances between the systemic and pulmonary circulations. Flow through the PDA is usually left to right, adding an additional volume burden to the lung that is already coping with RDS. Significant RDS can be accompanied by elevations in PVR that are high enough to cause bidirectional or even right-to-left shunting at the PDA. The elevated PVR also raises right heart pressures, causing the same phenomenon to occur at the foramen ovale. A patent foramen ovale (PFO) provides an opportunity for intracardiac right-to-left shunting. Any other type of lung disease (e.g., meconium aspiration or pneumonia) that is severe enough can also cause persistence of the fetal circulation. Nonsteroidal antiinflammatory drugs, via their antiprostaglandin action, can be used to induce closure of a PDA. Indomethacin is the preferred agent (Giroud and Jacobs, 2007).
Pulmonary Blood Flow and Pulmonary Vascular Resistance
At midgestation, PVR is estimated to be tenfold higher than it is 24 hours after an uncomplicated birth. During the last trimester, PVR decreases slightly to levels seven- to eightfold greater than it is 24 hours after delivery (Rudolph, 1979). This reduction results from the physical growth of the pulmonary vasculature that increases the cross-sectional area by more than the corresponding increase in blood flow. Yet, PVR still remains high immediately prior to birth and must fall dramatically in the first postnatal minutes to ensure survival. Pulmonary blood flow increases by an amount corresponding to the decrease in PVR, and pulmonary blood pressure falls by 50% (Fig. 4-7). Thus, the pulmonary vasculature presents a puzzle that flies in the face of the normal principles of cardiovascular embryology. The guiding principle of cardiac and vascular development is that once normal structures form, the correct pathway for blood flow is established. Once flow is established, normal growth ensues. The pulmonary vasculature must grow normally despite greatly reduced flow. The elevated PVR necessary for fetal existence cannot be solely the result of anatomic hypoplasia. If this were the case, the transition to postnatal life would be rocky indeed. Rather, the pulmonary vessels must be of normal size but retain the ability to markedly vasoconstrict. Even more remarkable is that the ductus arteriosus is an outgrowth of the pulmonary arterial system, yet it responds in a completely opposite fashion to the pulmonary vasculature. The very stimuli that raise PVR in utero cause dilation of the ductus arteriosus. The factors that govern pulmonary vasoreactivity have still not been fully elucidated, but our understanding has grown considerably in the last 20 years (Martin et al., 2006).
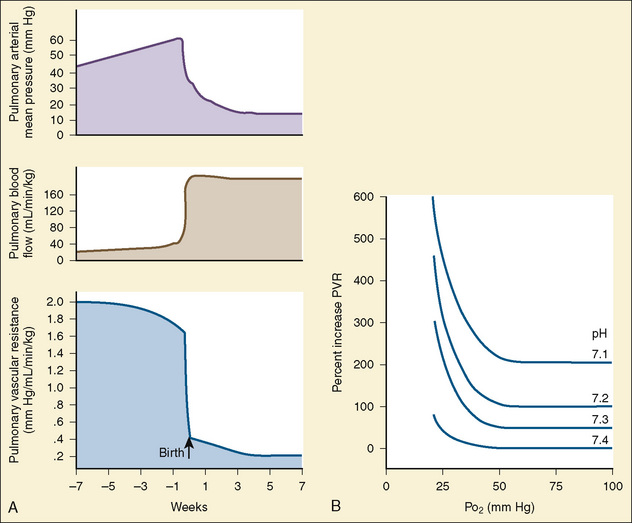
The primary force driving high fetal PVR is hypoxemia. The estimated Po2 in the pulmonary arteries is less than 20 mm Hg. This phenomenon of hypoxemia-induced pulmonary vasoconstriction (HPV) persists into adulthood, where it is vital in maintaining oxygen saturation during one-lung anesthesia. In utero, the mechanism of HPV is not fully understood. Oxygen is a potent stimulator of endothelial- derived vasodilating substances such as nitric oxide and prostacyclin. Nitric oxide activates soluble guanylate cyclase, which increases guanylate 3′-5′-cyclic monophosphate (cGMP). Prostacyclin stimulates adenylate cyclase to increase adenylate or adenosine 3′-5′-cyclic monophosphate (cAMP). Both cGMP and cAMP initiate vasodilation in pulmonary vessels. Under conditions of low oxygen tension, the release of nitric oxide and prostacyclin is presumably attenuated, tipping the balance in favor of vasoconstriction. The specific substances inducing pulmonary vasoconstriction are not well understood. Attention is focused on arachidonic acid and its metabolites and the potent vasoconstrictor endothelin (Tod and Cassin, 1984; Hickey et al., 1985a; Ivy et al., 1996).
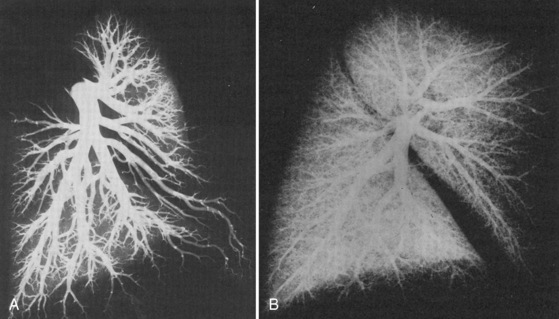
At delivery, mechanical and biochemical factors lead to the abrupt fall in PVR (Teitel et al., 1990). Aeration of the previously fluid-filled lungs removes the external compressive force on the pulmonary vasculature. Responding to the sudden rise in oxygen tension, the endothelium secretes potent vasodilators, nitric oxide and prostacyclin. Luminal diameter increases as endothelial and smooth muscle cells become thinner. The increase in blood flow further recruits small lumen vessels, leading to an overall increase in the cross-sectional area of the pulmonary vascular bed. Smooth muscle relaxation occurs in the larger pulmonary vessels. This first, rapid phase of pulmonary vasodilation is followed by a period of remodeling that lasts for months. During this time there is maturation of vascular smooth muscles with continuing modest declines in PVR. PVR approximates adult values by about 2 months of age, and the remodeling process is usually complete by 6 months of age. During the first few years of life, new vessels develop to supply the growing lung parenchyma (Fig. 4-8). It is important to understand that in the early postnatal period, PVR is markedly affected by hypoxia and acidosis (Rudolph and Yuan, 1966). More recently, pain has also been associated with increasing PVR.
Myocardial performance
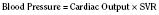
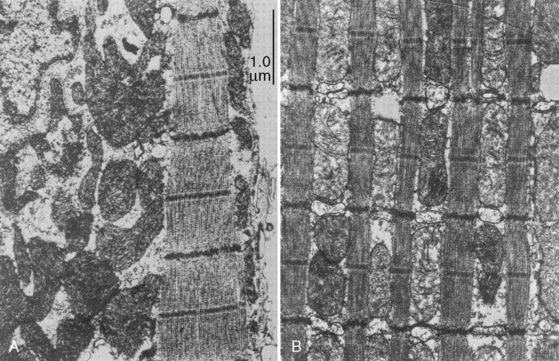
Cardiac cells are known as myocytes. Hyperplasia, which is an increase in cell number, is responsible for growth of the myocardium during fetal life. Hyperplasia continues into the early newborn period, after which increased demands on the heart can only be met by hypertrophy (increase in myocyte size). The functional unit of each myocyte is the myofibril. Myofibrils consist of contractile proteins arranged in repeating units called sarcomeres. The immature myocyte shape is rounded compared with the rodlike appearance of the adult myocyte. Immature myocytes also have a much larger surface-area to volume ratio. Adult myocytes contain multiple repeating rows of longitudinally arranged myofibrils. The newborn myocyte has fewer myofibrils in a more chaotic and scattered intracellular arrangement (Fig. 4-9). Sarcomere volume is only 30% of the newborn myocyte compared with 60% in the adult (Baum and Palmisano, 1997). The T-tubule system is a series of invaginations of the sarcolemma, or cell membrane, bringing it in close contact with the myofibrils. In this way, the action potential can rapidly disperse itself throughout the myocyte. Both the sarcolemma and T-tubule system are relatively well developed in the human newborn. The sarcoplasmic reticulum (SR) is a tubular network regulating the uptake, storage, and release of intracellular calcium. The adult heart relies on the SR to fully regulate calcium transport. Contrastingly, the newborn heart has an underdeveloped sarcoplasmic reticulum. There is more reliance on the sarcolemma and T-tubule system for the appropriate movement of calcium necessary for contraction and relaxation. The newborn has decreased contractile reserve on the basis of reduced sarcomere number and an immature system of calcium transport. The reduction in sarcomeres also reduces the compliance of the immature heart.
Preload
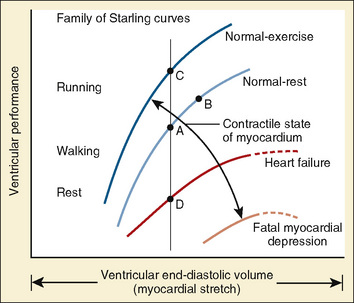
In 1895, the German physiologist Otto Frank published his observations on the relationship of diastolic filling of the heart and the pressure the heart was able to generate during systole (Frank, 1895). Ernest Starling, an English physiologist, conducted the classic experiment in the early 1900s that defined the length-tension relationship for cardiac muscle (Fig. 4-10). Understanding his experiment is important, because in the laboratory he was able to isolate that which is impossible to isolate in vivo. Using a fixed weight to keep afterload constant, he was able to prove that the tension developed was proportional to the length of the muscle strip prior to stimulation. The greater the length of the muscle strip, the greater the tension it was able to develop. The Frank-Starling mechanism, or Starling’s Law of the Heart, is taken from a famous lecture by Dr. Starling himself, in which he stated, “the energy of contraction, however measured, is a function of the length of the muscle fiber” (Starling, 1918).
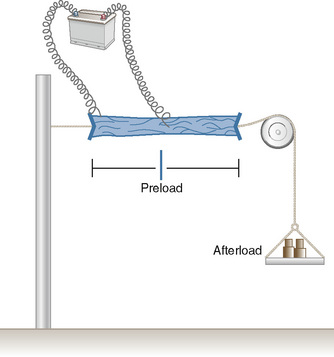
The clinical correlates of length and tension are left ventricular end-diastolic volume (LVEDV) and stroke volume, respectively. In the laboratory, the length of a muscle strip is easily determined because both ends can be fixed. This is very different from the intact heart, where the ventricle is a three-dimensional structure with complex geometry that defies the simple concept of length. Nevertheless, it is clinically appropriate to regard length as LVEDV. The LVEDV represents the loading of the ventricle. It is present before contraction, and therefore has come to be called preload. The force of ventricular contraction increases with increasing preload until a point is reached where the ventricle is over stretched and the force of contraction decreases. This point is the apex of the well-known Starling curve. Force of contraction is not synonymous with contractility, and for all points along a Starling curve, contractility is equal. Changes in the force of contraction, however, occur when preload changes (Fig. 4-11). Laboratory evidence has shown that resting sarcomere distance is 1.6 microns, with optimal conditions occurring at 2.2 microns. Excessive stretch, causing decreased force of contraction, does not occur until sarcomere length reaches 3.5 microns (Sonnenblick, 1974). In the laboratory the force of contraction increases until the sarcomere is stretched to over twice its resting length. If this translated fully to the intact heart, any increase in preload would result in increased force of contraction, because physiologically achieving a doubling of resting sarcomere length is almost impossible. However, well before a doubling of resting sarcomere length is reached, the patient falls on to the descending limb of the Starling curve. The reason is the relationship between volume and pressure, which is known as compliance.
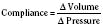
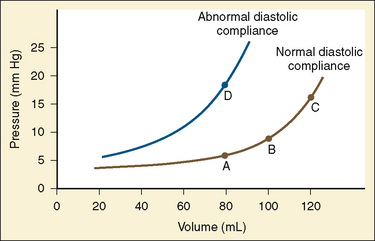
The change in pressure resulting from a change in volume is compliance. Volume is in the numerator of the equation meaning that if a large increase in volume is met by a relatively small rise in pressure, compliance is high. As a completely empty ventricle is filled, the initial rise in LVEDP is small. In this situation the ventricle is said to be compliant. When the ventricle is full, a small increase in volume results in a large increase in pressure. The ventricle is now poorly compliant or “stiff.” As with the Starling curve, it is important to note that moving to different points on the compliance curve does not mean that the intrinsic compliance of the ventricle has changed. A true intrinsic change in compliance will be reflected in a new compliance curve (Fig. 4-12). In adult cardiac medicine it is readily appreciated that ischemia, infarction, or hypertrophy result in a stiffer, or less compliant, ventricle. However, no ventricle, regardless of its intrinsic compliance, has an infinite ability to accept volume. Eventually the inflection point on the curve will be reached and pressure will climb rapidly for any given increase in volume. It is this sharp increase in pressure beyond the inflection point that is responsible for the descending limb of the Staring curve.

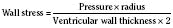
The response of the newborn heart to volume loading (relative insensitivity) has been the source of great confusion (Rudolph, 1974). This confusion stems from experimental work done in the 1970s and is the logical extension of the known structural differences in the newborn heart (Romero and Friedman, 1979; Gilbert, 1980). The response to preload was investigated in fetal sheep for the left and right ventricles. For either ventricle, isolated output rose only slightly with increases in filling pressures. Using a microsphere technique in fetal sheep, alterations in combined cardiac output were measured as blood volume was modulated (Gilbert, 1980). With a decrease of 10% in circulating volume, there was a significant drop in right atrial pressure, as well as in cardiac output. In contrast, cardiac output did not significantly change despite a significant change in right atrial pressure with a 10% increase in circulating blood volume from baseline. These data lead the author to conclude that the fetus operates at the upper end of the Starling curve and possesses limited cardiac reserve.
With only half the amount of sarcomere volume that is in an adult, connective tissue comprises a much greater percentage of the newborn heart. The myofibrils are fewer in number with a disorderly arrangement in the myocyte. There is much more stiff connective tissue. The sum total of these changes makes it plausible that the relatively noncompliant newborn heart does function at the upper end of its Starling curve, where the response to volume loading is blunted. In fact, the original experiments failed to account for an inevitable consequence of increases in preload. An increase in the preload leads to greater stroke volume, and if heart rate is unchanged, cardiac output must increase as well. Blood pressure must now rise unless there is a corresponding drop in SVR. Therefore, an increase in preload leads to an increase in afterload, which then acts to reduce stroke volume. This created the impression that the newborn heart could not increase stroke volume in response to volume loading. Other work, controlling for arterial pressure, shows that the newborn heart is indeed responsive to volume within the limitations of its decreased compliance (Fig. 4-13) (Kirkpatrick et al.; 1976, Hawkins et al., 1989). This has been shown in the laboratory with a sheep model and though echocardiography in the human fetus. The newborn can be likened to the hypertensive adult with diastolic dysfunction. This is not to suggest that the otherwise healthy newborn has diastolic dysfunction, because the newborn heart is not dysfunctional but normal for its stage of development. Rather, the lesson of diastolic dysfunction is that the ventricle is preload dependent. At the low end of the Starling curve, reduced preload is poorly tolerated. The upper end of the Starling curve is flattened, reflecting its reduced compliance. Between these two points is the steepest part of the curve, where increases in LVEDV result in significantly greater stroke volume. Clinical experience observing the response to volume infusion confirms this property of the newborn heart.
Afterload
In addition to preload, there is another loading force that influences myocardial performance. The force that resists the ejection of blood is known as afterload. In isolated muscle strip experiments, the afterload is represented by the weight against which the muscle contracts or shortens. The stretch applied to the isolated muscle strip before contraction is fixed, allowing the experiment to be conducted at a constant length, or preload. Plotting the velocity of myocardial shortening against progressively increasing afterload reveals an inverse relationship. The shape of the curve is exponential with the points of maximal and zero shortening both occurring at physiologically impossible limits. The greatest shortening velocity occurs at zero load. When the load is so great that no muscle fiber shortening can occur, isometric contraction occurs. Between these two extremes, it is intuitive that increasing afterload results in reduced velocity of muscle fiber shortening. Transferring this straightforward laboratory concept to the intact circulation is difficult (Martin et al., 2006). The force generated by the shortening of an isolated muscle strip is directed in only one plane. This is very different from the three-dimensional contraction of the left ventricle with its asymmetric geometry. Additionally, in the laboratory the force that resists the shortening of the muscle is the weight applied to the muscle strip. Physiologically, the force that resists the ejection of blood is a complex interplay between blood pressure, vascular impedance, walls stress, and inertia. Finally, blood pressure and SVR are commonly used measures of afterload, but neither is fully accurate.
These two hypothetical patients are very different, yet the SVR is similar. The first patient has normal cardiovascular function and the second patient’s cardiovascular function is compromised. A reduction in contractility has caused a fall in cardiac output despite the attempts of the ventricle to compensate with a higher diastolic filling pressure. This example demonstrates how SVR in isolation does not necessarily reflect large changes in loading conditions and cardiac output. The modified Law of Laplace describes a better approximation of afterload (see Preload section p. 91). As discussed in the section on preload, wall stress was demonstrated to be a key component of myocardial oxygen demand. During left ventricular ejection, the value for pressure is the systolic blood pressure. By including the radius and accounting for the compensatory mechanism of left ventricular hypertrophy the modified Law of Laplace provides a more comprehensive view of the forces that resist ventricular ejection. Importantly, it demonstrates the concept that the dilated ventricle with an increased radius must eject blood under increased afterload conditions. Given the inability to easily measure ventricular radius and thickness, the modified Law of Laplace is not a clinical parameter to guide management. It is presented here to help the reader conceptualize afterload. It still cannot describe all the variables that account for afterload in the intact cardiovascular system.
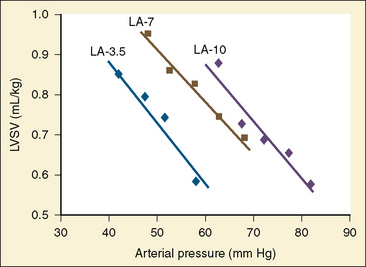
Studies of the intact heart that assess the effect of changes in afterload on cardiac output are difficult to interpret without controlling for the other variables. The most important additional variable is preload. Preload and afterload are linked. For instance, pharmacologically increasing afterload decreases stroke volume. As the heart ejects less blood with each contraction, the end-diastolic volume must increase if venous retum is held constant. With an increase in LVEDV the Starling curve dictates an enhanced ventricular force of contraction, which acts to return stroke volume to its previous level. The general inverse relationship between afterload and stroke volume holds across the spectrum from fetal life to adulthood (Friedman, 1972). The question is whether the immature myocardium is excessively sensitive to increases in afterload. In the laboratory, afterload can be assessed while strictly controlling for preload. These studies have revealed that the fetal myocardium is indeed more sensitive to increases in afterload than the adult myocardium (Friedman, 1972). Animal experiments with an intact circulation also show a decrease in ventricular output under conditions of increasing afterload (Fig. 4-13) (Gilbert, 1982; Thornburg and Morton, 1986). Van Hare and colleagues (1990) used a balloon occluder in the aortas of fetal sheep to modulate afterload while keeping preload constant. Combined ventricular output was measured by transducing aortic flow. They noted that at physiologic mean atrial pressures, there was an inverse linear relationship between mean arterial pressure and stroke volume. This exists for both right and left ventricles. Despite this, a fetus normally makes a smooth transition to postnatal life when the low-resistance placenta is removed. At birth, pulmonary blood flow dramatically increases, raising left-ventricular preload. Stroke volume increases, which leads to an increase in systemic blood pressure. Despite the rise in blood pressure and the loss of the low-resistance placenta, cardiac performance in the newborn is not compromised. The explanation is that much of the rise in afterload is secondary to increased preload. In clinical practice with newborns and young children, it is rare to encounter conditions of purely increased afterload. A more likely situation is significant hypovolemia that results in hypotension. Despite the low afterload state, overall cardiac performance is impaired, because the decrease in afterload is caused by decreased preload. The correct therapy is to restore intravascular volume. Blood pressure improves, and the increased afterload is well tolerated. This example illustrates the interrelationship between these two variables of cardiac output.
It has been shown that when preload is held relatively constant, cardiac output moves inversely within the physiologic range of afterload. The relevance of afterload in the newborn stage might be questioned, because systemic hypertension is so rare. The right ventricle develops in an environment of raised PVR, yet after birth it is more sensitive to increases in afterload. Although there is limited ability of the systemic circulation to become hypertensive, the pulmonary circulation, under appropriately provocative conditions, can revert to its fetal state. Postnatally, the right ventricle begins to remodel in response to decreasing pulmonary artery pressures. Sustained elevations in pulmonary artery pressures decrease right ventricular output and reduce left-ventricular preload (Thornburg and Morton, 1983). Because both ventricles share the septum, the strain on the right ventricle impairs left-ventricular filling and contraction (Rein et al., 1987). Reductions in left-sided preload and contractile force demonstrate how the inability of the right ventricle to cope with increased afterload can lead to decreases in systemic cardiac output. In the newborn stage, the potential for pulmonary hypertension far exceeds that of systemic hypertension. This is the most common scenario for a significant rise in afterload while preload remains relatively unchanged. The strain on the right side of the heart and ventricular interdependence may lead to biventricular failure.
Contractility
The immature myocardium has a reduced sarcomere concentration combined with a transport system for calcium that is not fully developed. The myofibrillar arrangement is also more disorganized in the infant heart when compared with the adult heart. Mitochondria, the energy powerhouse of the myocyte, are reduced in number in the immature heart (Barth et al., 1992). Based on the above differences, a reduction in contractility is expected. Contractility is measured by the tension developed in the isolated muscle strip. This expected reduction in contractility of the immature myocardium is confirmed in laboratory experiments where preload and afterload are controlled. Across the entire range of loading conditions, fetal cardiac muscle generates less tension than adult myocardium (Fig. 4-14) (Friedman, 1972; Romero et al., 1972). In an intact heart it is not possible to fully separate the influences of different loading conditions. Nevertheless, fractional area shortening of the ventricle measured echocardiographically is used as a measure of contractility. Decreased fractional area shortening has been observed in the fetal heart (St. John Sutton et al., 1984). Effecting a true increase in contractility requires either a greater number of contractile elements or more vigorous action by the contractile elements already present. Increasing the number of contractile elements (hyperplasia and hypertrophy) occurs as a normal part of development. Improving the action of the contractile elements already present requires an understanding of the singular role of calcium. Therapy to improve inotropy exerts its action at the myocyte level by affecting the levels of intracellular calcium.
Calcium and Diastolic Function
A comprehensive understanding of the excitation-contraction coupling mechanism of contraction and relaxation is important for the safe practice of pediatric anesthesia. The relevant points are included here. The formation of actin-myosin cross-bridges occurs under the influence of calcium. Tropomyosin and troponin are inhibitory proteins that prevent the formation of actin-myosin cross-bridges. Calcium induces conformational changes in tropomyosin and troponin that remove their inhibition to actin-myosin cross-bridge formation. This is the basis of contraction and requires major changes to intracellular calcium content. It is estimated that the difference between diastolic and systolic calcium levels is 100-fold (10–7 M to 10–5 M) (Shah et al., 1994). Responsibility for these large changes in intracellular calcium concentration lies with the integrated function of the sarcolemma, the T-tubule system, and the SR. Breaking the actin-myosin cross-bridges and returning the ventricle to its baseline state is an energy-consuming process that requires adenosine triphosphate (ATP) and calcium reuptake. This is primarily accomplished by the removal of calcium into the SR and the sarcolemmal Na+–Ca2+ exchanger (Mahony, 2007). Ryanodine is an inhibitor of SR function. In the presence of ryanodine, fetal and newborn hearts are minimally affected, whereas adult hearts suffer a significant decline in contractility (Penefsky, 1974).
Experimental modulation of calcium handling has been shown to alter ventricular mechanics. In a study with mice that overexpressed the Ca2+ ATPase, the rate of myocardial relaxation was directly correlated with the rate of calcium uptake by the SR (He et al., 1997). In guinea pigs, ryanodine blockade of SR function caused impaired relaxation (Kaufman et al., 1990). Additionally, there were age-dependent changes in the relaxation response. In adult hearts, ryanodine blockade produced a greater impairment of relaxation compared with immature hearts. Concomitantly, there was greater density of Ca2+ pumps, greater calcium-dependent ATPase activity, and greater uptake of calcium in isolated SR vesicles from adult hearts as compared with those isolated from immature hearts. With decreased SR function in the immature heart, the extrusion of calcium across the cell membrane assumes greater importance. The Na+–Ca2+ exchanger provides the primary mechanism for this. The exchanger is sensitive to membrane potential as it exchanges three sodium ions for one calcium ion. Developmental changes in the exchanger function have been demonstrated, and there are differences among species in relative function (Nakanishi and Jarmakani, 1981; Artman, 1992; Boerth et al., 1994). In the rabbit, with its poorly developed SR exchanger, mRNA is significantly elevated in the neonate versus the adult (Artman, 1992). The relative contribution of calcium sequestration between the SR and the exchanger in the human is similar to that of the rabbit (Mahony, 2007). In addition to reduced calcium handling, there may be developmental changes in troponin interactions that effect calcium binding. There is differential expression of cardiac and slow skeletal-muscle isoforms. In fetal hearts, the predominant form is the slow-skeletal form. Shifts to the cardiac isoform are completed in the first year of life. This is significant in that the cardiac isoform has a decreased affinity for calcium when phosphorylated, potentially aiding the removal of calcium from troponin C.
The sum total of these events is to demonstrate that across all age ranges the intracellular handling of calcium is critical for systolic function and even more important for diastolic function. The diastolic characteristics of the immature myocardium have been studied. The immature myocardium has been described as “stiffer” when compared with the adult myocardium. Ventricular compliance increases with maturation (Friedman, 1972; Romero et al., 1972; Kaufman et al., 1990). Measurements of rates of pressure change demonstrate decreased relaxation capabilities in neonatal hearts when compared with adult hearts (Palmisano et al., 1994). There are a number of potential factors involved. Structural and contractile protein changes, extra-cardiac structures, and maturing organelle function have been studied. The diastolic relaxation properties of the ventricle are a key determinant of the ventricle’s compliance. Reductions in calcium reuptake lead to expected decreases in diastolic relaxation (Kaufman et al., 1990). Echocardiographic studies on human fetuses with normal hearts have demonstrated age-related changes in early diastolic flow that are consistent with improved relaxation (Kenny et al., 1986; Harada et al., 1997). Consequently, intracellular calcium homeostasis in the newborn is more dependent on a normal serum ionized calcium level, and the newborn tolerates hypocalcemia poorly. Immaturity of calcium transport leads to a decrease in systolic-force generation and a decrease in diastolic relaxation. The manifestation of impaired diastolic relaxation is a reduction in compliance. Clinically, these issues must be appreciated when interpreting assessments of volume based on pressure readings such as the central venous pressure (CVP).
Heart Rate
The question is whether the newborn who has a heart rate within normal range is fundamentally different than the adult. When corrected for weight, stroke volume is similar across all ages. The high cardiac output of newborns and infants can only be achieved with a heart rate that is significantly higher than in adults. This has created the idea that the newborn is dependent on heart rate. Here it becomes difficult to sort out the isolated effect of heart rate from its effects on loading conditions and contractility. It is not intuitive as to why changes in heart rate alter contractility. The mechanism is known as the force-frequency relationship. Experiments using atrial pacing, while controlling loading conditions, demonstrate an increase in stroke volume with an increase in heart rate (Anderson et al., 1982).With constant loading conditions, the only explanation for increased stroke volume is that contractility has increased. Thus, an increased heart rate improves contractility. The basis for the force-frequency relationship is that an increase in heart rate is accompanied by enhanced release of intracellular calcium (Parilak et al., 2009). There is a suggestion that the force-frequency relationship has minimal effect in newborns but is present during infancy (Wiegerinck et al., 2008). Spontaneous increases in heart rate also improve cardiac output. In this scenario, the increase in heart rate is the result of a neurohumoral stimulus with an effect that cannot be isolated to producing tachycardia. Both preload and contractility can be expected to increase, as long as the increase in heart rate does not lead to deleterious changes in preload or myocardial oxygen supply and demand. The combination of these factors allows the newborn and infant to use heart rate to significantly augment cardiac output.
Integrating Preload, Afterload, and Contractility
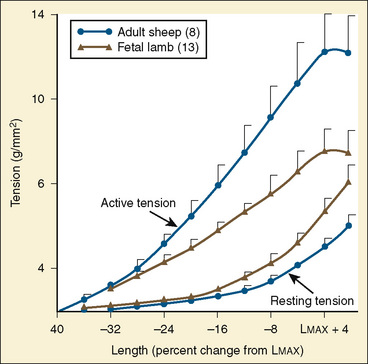
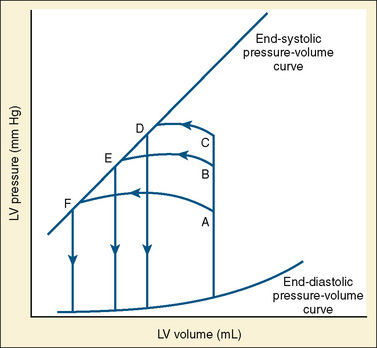
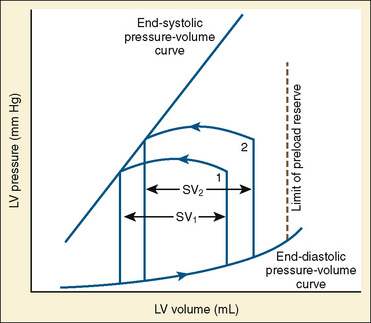
The determinants of myocardial performance, while discussed in isolation, are actually intricately linked. Diagrammatically, the determinants of myocardial performance can be represented by ventricular pressure-volume loops (Suga et al., 1973). The loop shows the pressure and ventricular volume changes that occur during one cardiac cycle. Increases in preload while afterload is held constant result in greater stroke volume (Fig. 4-15). This simple curve does not provide any information about contractility. The end systolic pressure-volume relationship (ESPVR) is a family of curves that is generated by rapidly altering preload or afterload. The curves create a series of points that are connected to become the ESPVR line. The slope of this line represents contractility (Fig. 4-16). The pressure-volume loop illustrates the concept that movement along the ESPVR line reflects changes in loading conditions while contractility remains constant. In Figure 4-16, the increases in afterload result in a series of points that are connected to become the ESPVR line. The slope of the ESPVR line represents contractility. The two figures of ventricular pressure-volume loops represent an idealized situation where preload and afterload can be manipulated independent of each other. In the intact organism, preload and afterload are linked. The interdependence of preload and afterload is shown in a new ventricular pressure-volume loop (Fig. 4-17).
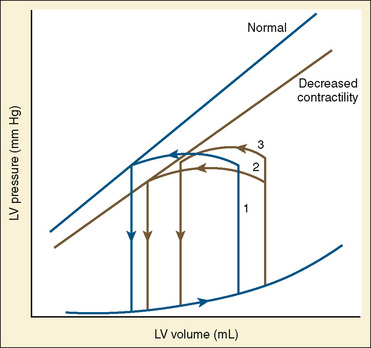
In states of decreased contractility, the ventricular pressure-volume loop displays the limitations of the failing heart. Analysis of the ventricular pressure-volume loop shows that the failing ventricle is sensitive to changes in loading conditions (Fig. 4-18). The ESPVR line has shifted down and to the right. With preload held constant, stroke volume is reduced. The body’s response to this situation is to increase LVEDV in an attempt to preserve stroke volume. Stroke volume has improved at the expense of a higher end-diastolic pressure. The ventricular pressure-volume loop demonstrates why afterload reduction is the cornerstone of medical therapy for the failing heart. Any increases in afterload come at the expense of stroke volume, with a limited ability for compensatory changes in preload.
Developmental aspects of cardiomyocyte structure and function
Subcellular Structures
Sarcolemmal Ion Channels
Numerous voltage-dependent and ligand-gated ion channels reside in the sarcolemma, and a full discussion far exceeds the space available in this chapter. Although developmental changes in the inward sodium current have been noted in several species, there appear to be few developmental changes in the human atrium (Sakakibara et al., 1992). A variety of inward and outward potassium channels exists. Although developmental changes have been documented, methodologic differences and differences among species do not allow application to human development (Sanchez-Chapula et al., 1994; Xie et al., 1997; Morrissey et al., 2005). Na+,K+ ATPase maintains the sodium gradient across the cell and is inhibited by the cardiac glycosides, such as digitalis. There are developmental changes in the isoform distribution of the enzyme subunits, and this enzyme has less activity in immature myocardium. Calcium handling is crucial to myocardial contraction. Calcium channel (Ica) density increases two to threefold in the developing rabbit, although the voltage sensitive activation is similar to that of humans (Osaka, 1991; Huynh et al., 1992; Wetzel et al., 1993). In one study of human atrial myocytes (presumably from ill children), younger hearts had decreased calcium channel density, and in another study, more rapid inactivation of calcium current was evident (Hatem et al., 1995; Roca et al., 1996). However, in the realm of human myocardial development, many of these hearts were fairly mature when studied. The Na+–Ca+ exchanger, which can serve to bring calcium either into or out of the cell, has higher activity in immature myocardium in a variety of species (Artman et al., 1995). This is a purported source of additional calcium entry into the contractile apparatus in immature myocardial cells that have relatively deficient sarcoplasmic reticulum. There is increased activity of the Na+–H+ exchanger in immature myocardium, and this has been implicated as a factor in the greater resistance of immature myocardium to acidosis (Downing et al., 1966; Haworth et al., 1997).









