Sites of action of antiarrhythmic drugs
| Class I |
IA
|
IB
|
IC
|
Class II
|
Class III
|
Class IV
|
Class I
Class I drugs work in a similar way to local anaesthetic agents (lidocaine is used for both roles). They act by slowing sodium entry into cells through the fast voltage-gated sodium channels that primarily affect the non-nodal areas characterised by a fast depolarisation action potential. They reduce the maximum rate of rise of phase 0 depolarisation. The rate of phase 4 sinoatrial node depolarisation may also be reduced, and with it spontaneous automaticity.
Fast voltage-gated sodium channels may exist in three states – resting, open and refractory. In normal myocardium their state switches between resting and open, but ischaemia results in prolonged depolarisation and the channel becomes refractory. The class I antiarrhythmics block open channels so that the more frequent the action potentials the more ionophores become blocked. The first action potential shows a slight reduction in phase 0 depolarisation. Subsequent action potentials show a progressive reduction in the rate of depolarisation as more channels are blocked. The block is therefore use-dependent.
Class I has three subdivisions, based on the effect on the action potential duration. The effect on action potential duration is related to the type of sodium channel affected, as follows:
The key features of the subdivisions of class I antiarrhythmics are listed in Figure 36.3, and their ECG effects are shown in Figure 36.4.
Class IB antiarrhythmics have a receptor association/dissociation cycle shorter than the cardiac cycle. The initial drug effect causes gradual blockade of the ionophores to develop during the action potential depolarisation. By the time the next action potential occurs, however, the drug will have dissociated from its receptor again. If a premature depolarisation (e.g. a ventricular premature beat) occurs then the myofibril will still be blocked. Class IB antiarrhythmics bind preferentially to refractory channels, and are therefore selective for ischaemic myocardium. Class IB drugs also elevate the fibrillation threshold. Lidocaine is discussed in Chapter 33. Mexiletine is similar but also has anticonvulsant properties, possibly due to GABA reuptake inhibition.
Class IC drugs have a much slower association/dissociation cycle, lasting longer than the cardiac cycle, so the block is relatively constant from cycle to cycle. The overall effect is a general reduction in excitability, and this is therefore more suitable for re-entrant-type rhythms. There is little selectivity for the refractory channels of ischaemic myocardium.
Class IA drugs have features midway between those of IB and IC.
Class IA antiarrhythmics are primarily used for supraventricular tachycardias, and class IB and C for ventricular tachycardias.
| Class | Example | dV/dt of phase 0 | Repolarisation | PR | QRS | QT |
|---|---|---|---|---|---|---|
| IA | Quinidine | Slowed | Prolonged | ↑ | ↑ | ↑ |
| IB | Lidocaine | Little effect | Shortened | 0 | 0 | ↓ |
| IC | Flecainide | Marked slowing | Little effect | ↑↑ | ↑↑ | 0 |
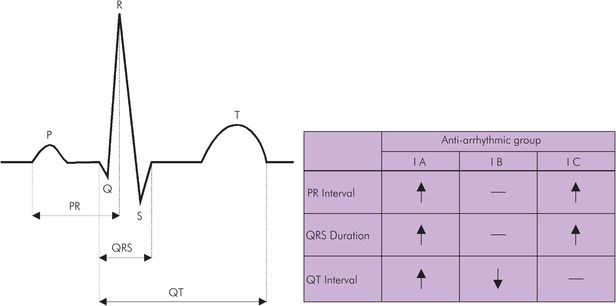
ECG effects of antiarrhythmic drugs
Class II
Class II antiarrhythmics (β-blockers) competitively block the effects of circulating and neurotransmitter catecholamines, reducing arrhythmogenicity, heart rate and contractility. This slows and so lengthens phase 4 depolarisation, the phase which is shortened by catecholamines. The action potential is shortened and the refractory period of the atrioventricular (AV) node is prolonged. These Gs-coupled effects are mediated by a slow L-type Ca channel influx that may be particularly useful in preventing post-myocardial infarction ventricular arrhythmias, when catecholamine levels may be high. Some potassium, chloride and even sodium channel modulation may also be involved. β-blockers are covered in detail in Chapter 35.
Class III
Class III drugs (e.g. amiodarone, bretylium, sotalol) prolong repolarisation and therefore the action potential, and increase the effective refractory period. Their principal uses are for the following arrhythmias:
Atrial tachycardia
Atrial flutter and fibrillation
Re-entrant junctional tachycardia
Ventricular tachycardia
Amiodarone is particularly effective for the treatment of atrial tachyarrhythmias when Wolff–Parkinson–White syndrome is also present. At higher doses, the effect may be similar to β-blockade and there is a quinidine-like action. Marked hypotension may result from IV administration.
Bretylium tosylate is reserved for use in resuscitation. It is given intravenously or intramuscularly in the treatment of ventricular fibrillation after DC shock and lidocaine have been tried. It accumulates in sympathetic ganglia and reduces noradrenaline release. There is an initial postganglionic discharge. It may cause hypotension, nausea and vomiting, and should be used with caution when a cardiac glycoside has been previously administered.
Sotalol is a β-adrenoceptor antagonist and is classified as both a class II and a class III antiarrhythmic. Sotalol causes both β1 and β2 antagonism (mainly by the l isomer). Its place amongst the class III antiarrhythmics is due to K+ channel inhibition, which is equally attributable to both stereoisomers. It is indicated for the treatment of paroxysmal supraventricular tachycardia, ventricular premature beats and ventricular tachycardia. It is more suitable than lidocaine for the treatment of spontaneous sustained ventricular tachycardia secondary to coronary disease or cardiomyopathy. The usual precautions for β-blocker therapy apply. Use with caution when there is hypokalaemia and when other agents that prolong the QT interval are concurrently administered.
Class IV
There are numerous calcium antagonists (see below), having different sites of action. Verapamil and diltiazem have antiarrhythmic activity, but nifedipine does not. Slowing of calcium influx reduces the duration of phases 2 and 3 of the action potential. At the AV node, this action is particularly beneficial in preventing re-entry rhythm problems.
Adenosine
Adenosine is used for rapid conversion of paroxysmal supraventricular tachycardias back to sinus rhythm (including Wolff–Parkinson–White syndrome). It may also be used in the diagnosis of conduction defects. Adenosine is ubiquitous in the body, in combination with phosphate (e.g. cAMP) and has predominantly inhibitory effects. There are four specific types of adenosine receptor, A1, A2A, A2B and A3, the effects of which are detailed in Figure 36.5.
A1
|
A2A
|
A2B
|
A3
|
The effect of adenosine in causing transient slowing of AV nodal conduction allows normal sinus nodal discharge to initiate a normal pattern of electrical depolarisation throughout the heart, and so normal sinus rhythm resumes, before the drug has been eliminated. This appears to be mediated by increasing myocardial K+ flux. The A1 purinoceptors are linked by a stimulatory G protein to the same transmembrane K+ channels that are opened by M2-muscarinic acetylcholine receptors. It is an unusual drug in that a key to successful use is that its IV injection must be rapid. This is because it is metabolised rapidly, with a half-life of only 8–10 seconds. Unlike verapamil, it can be used safely in conjunction with β-blockers, but is contraindicated in asthma, sick sinus syndrome, and second- and third-degree AV heart block.
Cardiac glycosides
The cardiac glycosides are a group of naturally occurring compounds used to improve myocardial contractility and reduce cardiac conductivity. Cardiac glycosides are found mainly in three botanical species, white foxglove (Digitalis lanata), purple foxglove (D. purpurea) and climbing oleander (Strophanthus gratus).
Chemistry
The cardiac glycosides share the same basic structure:
Steroid nucleus (cyclopentanophenanthrene)
Lactone ring (five- or six-membered) – the aglycone
Carbohydrate (up to four monosaccharide units)
The carbohydrate moiety is responsible for solubility, and the lactone ring confers pharmacological activity. Saturation of the lactone rings reduces potency, and opening of the ring abolishes the pharmacological activity. Only rings A and D of the steroid nucleus are coplanar, whereas in adrenal steroids rings A and C, and B and D are coplanar.
Pharmacological effects
Cardiac glycosides increase myocardial contractility and slow conduction at the AV node. Isometric and isotonic contraction of both atrial and ventricular muscle is improved. Cardiac glycosides cause directly mediated vasoconstriction.
Clinical uses
Cardiac glycosides are used to improve contractility in hypervolaemic myocardial failure. In chronic atrial fibrillation ventricular rate becomes slower because of a direct reduction in AV nodal conduction and by a secondary reduction in vagal tone.
Mechanisms of action
Sodium–potassium adenosine triphosphatase (Na+K+ATPase)
Cardiac glycosides specifically and reversibly bind to cardiac cell membrane Na+K+ATPase, which alters the electrolyte balance inside the myocardial fibres with more sodium and less potassium intracellularly. This results in a reduction of the transport of sodium into the cell using the Na+Ca2+ ion exchange system, and intracellular calcium is better maintained and increased. The increased calcium may be responsible for the positive inotropy. However, at higher doses this interference with an essential membrane pump may be responsible for toxicity.
Interference with neuronal catecholamine reuptake
At low biophase concentrations, there is an increase in local catecholamine levels due to interference with neuronal catecholamine reuptake. This has positive inotropic and chronotropic effects, and may in fact increase overall Na+K+ATPase activity. There is also an increase in local acetylcholine levels, resulting in negative inotropic and chronotropic effects.
Elimination
The glycosides are highly bound to cardiac muscle, and therefore have very high volumes of distribution. This slows elimination considerably, as most are excreted unchanged in the urine, both by simple filtration and also by tubular secretion. Digitoxin is the exception, being primarily metabolised by hepatic microsomal enzymes.
Toxicity
The cardiac glycosides have a very low therapeutic index. Most of the toxic effects are due to potassium loss. It is therefore particularly important to consider potassium-sparing diuretics when diuretics are needed in conjunction with cardiac glycosides. Side effects of the cardiac glycosides are listed in Figure 36.6.
| Heart block |
| Cardiac arrhythmias |
| Fatigue |
| Nausea |
| Anorexia |
| Xanthopsia |
| Confusion |
| Neuralgia |
| Gynaecomastia |
Magnesium
Magnesium is the fourth most prevalent cation in the body, and the second intracellularly. Of total body magnesium, 53% is in bone, with 27% in muscle and only 0.3% in plasma. Its physiological roles include incorporation as a cofactor in over 300 enzyme systems, inhibition of IP3-gated calcium channels, muscle contraction, neuronal activity, neurotransmitter release and adenylyl cyclase regulation.
Pharmacologically, magnesium is used as replacement therapy. Magnesium deficiency is not uncommon in hospital patients, and in particular two-thirds of intensive care patients may be magnesium-depleted. Magnesium is effective in the treatment of eclampsia and pre-eclampsia and in preventing the hypertensive response to intubation. The treatment of cardiac dysrhythmias is particularly indicated if hypokalaemia coexists. A list of possible uses is given in Figure 36.7.
| Correction of magnesium deficiency |
| Control of eclampsia and pre-eclampsia |
| Inhibition of premature labour |
| Improvement of cardiac contractility |
| Limitation of myocardial infarct size |
| Component of cardioplegia (a mixture to arrest the heart during cardiac surgery) |
| Correction of cardiac dysrhythmias: emergency treatment of torsades de pointes, digoxin toxicity, life-threatening atrial and ventricular dysrhythmias |
| Prevention of hypertensive response to intubation |
| Reduction of catecholamine release in phaeochromocytoma surgery |
| Treatment of asthma |
Calcium antagonists
Calcium is involved not only in muscle contraction but also in neurotransmitter release, hormone secretion, platelet aggregation and enzyme function. There are numerous calcium channels across cell and other membranes. These may be active or passive, triggered by chemical mediators or voltage changes, and may be coupled with other ionic exchange. The term calcium channel blocker is generally used to describe those agents with a role in cardiovascular manipulation. These act primarily on the voltage-gated (L-type) calcium channels, preventing opening of the ionophore. There are three main groups (papaverines, benzothiazepines, dihydropyridines), each having characteristic actions. However, even within these groups, there is considerable difference between the individual chemical structures of the drugs, and in their calcium-channel specificity. The myocardial depression caused by the volatile anaesthetic agents halothane, enflurane and isoflurane is also the result of an alteration in calcium flux. Figure 36.8 shows the relative potencies of the three representative calcium antagonists.
Related posts:






Full access? Get Clinical Tree


