Cardiovascular
7-1 Acute Myocardial Infarction
Ralph Weiche
Clinical Presentation
Patients with acute myocardial infarction (AMI) present with chest discomfort, tightness, pressure, or a “squeezing sensation” located over the anterior chest. The chest pain may radiate to the jaw, neck, arms, back, or epigastrium. Associated symptoms may include dyspnea, nausea, anxiety, lightheadedness, syncope, or diaphoresis. Elderly patients, women, and patients with diabetes are more likely to have atypical presentations.
Pathophysiology
The term acute myocardial infarction refers to myocardial necrosis secondary to ischemia. Ischemia typically results from coronary artery atherosclerotic plaque rupture, with thrombus formation and consequent occlusion of the blood supply in a coronary vessel. Ischemia may also result from coronary artery vasospasm. In some patients, this vasospasm results from the use of sympathomimetic or serotonergic medications (e.g., cocaine, amphetamines).
Diagnosis
The diagnosis requires a high index of suspicion in patients with historical risk factors for coronary artery disease (smoking, age greater than 55 years, hypertension, diabetes, family history) and consistent symptoms. Patients may present with an absence of chest pain (“silent MI”), with an atypical history, or before visible changes in the electrocardiograph (ECG) or serum markers.
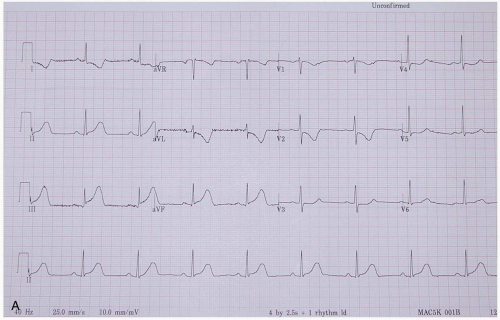 FIGURE 7-1 A: Inferior wall myocardial infarction. (Courtesy of Robert Hendrickson, MD.) |
ECG manifestations of AMI include sinus tachycardia (ST)-segment elevation greater than 1 mm in two contiguous leads and new Q waves.1 Myocardial ischemia may produce ST-segment depression or T-wave inversion. However, normal or nonspecific findings on ECG do not exclude the diagnosis of AMI, and ECG changes may be late findings.1
The anatomic localization of an AMI can be inferred by the distribution of ECG abnormalities: inferior wall (II, III, aVF), lateral wall (I, aVL, V4-V6), anteroseptal (V1-V3), anterolateral (V1-V6), right ventricular (RV4, RV5), and posterior wall (R/S ratio >1 in V1 and V2; upright T-waves in V1, V8, and V9).
Clinical Complications
Complications include systolic or diastolic dysfunction, decreased ejection fraction, cardiogenic shock, arrhythmias, valvular dysfunction (rupture of a papillary muscle or chordae tendineae), and septal or ventricular wall rupture.
Management
The management of AMI is complex, and as the state of the art evolves, so do the standards of care for AMI. Published guidelines should be reviewed for details.1 The initial management of suspected AMI should include adequate intravenous (IV) access, oxygen, cardiac monitoring, and placement in a location with defibrillation and cardiac medications readily available.1 All patients
should be treated with aspirin (unless allergic) and β-blockade (unless bradycardic, hypotensive, or intoxicated with sympathomimetics). Chest pain may be relieved with nitroglycerin or morphine sulfate. Patients should be evaluated with an ECG and laboratory values (myoglobin, creatine phosphokinase [CPK], troponin, or some combination of these). Reperfusion therapy with cardiac catheterization (if available) or fibrinolytics or both should proceed immediately on the diagnosis of AMI.1 Patients with an AMI should be placed in an intensive care setting, and on-site consultation with a cardiologist should be obtained.
should be treated with aspirin (unless allergic) and β-blockade (unless bradycardic, hypotensive, or intoxicated with sympathomimetics). Chest pain may be relieved with nitroglycerin or morphine sulfate. Patients should be evaluated with an ECG and laboratory values (myoglobin, creatine phosphokinase [CPK], troponin, or some combination of these). Reperfusion therapy with cardiac catheterization (if available) or fibrinolytics or both should proceed immediately on the diagnosis of AMI.1 Patients with an AMI should be placed in an intensive care setting, and on-site consultation with a cardiologist should be obtained.
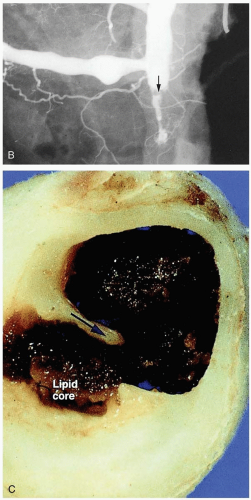 FIGURE 7-1, cont’d. B: The occluded artery (arrow) shows a thread of angiographic medium passing some distance into the thrombus, but there is no distal artery filling. C: The end of the torn plaque cap is visible (arrow); thrombus within the lipid core is in continuity with thrombus within the arterial lumen. (B and C, from Davies, with permission.) |
REFERENCES
1. Ryan TJ, Antman EM, Brooks NH, et al. 1999 update: ACC/AHA guidelines for the management of patients with acute myocardial infarction. Circulation 1999;100:1016-1030.
7-2 Unstable Angina
Todd McGrath
Clinical Presentation
Patients may present with substernal chest pain with or without radiation to the left arm, worsening pain similar to previous angina, resting chest pain, congestive heart failure (CHF), nausea, diaphoresis, and shortness of breath.1 Patients may have the following risk factors: age older than 70 years, diabetes, cigarette smoking, peripheral vascular disease (PVD), coronary artery disease, and male gender.1
Pathophysiology
Unstable angina refers to chest pain of cardiac origin that is either increasing in intensity and frequency or occurring without inciting events (i.e., pain at rest).
The cause of unstable angina is ischemia of the cardiac myocytes, which results from thromboembolization within the coronary arteries, most commonly due to rupture of atherosclerotic plaques.2
Diagnosis
The diagnosis is made by recognition of the clinical history and confirmed with an electrocardiograph (ECG) and cardiac biomarkers. Sinus tachycardia (ST)-segment depression or elevation or the presence of T-wave changes is sufficient to confirm the diagnosis of unstable angina.1 In patients with unchanged ECGs, biomarkers may reveal evidence of ischemia/unstable angina. Troponin T and I are highly specific for cardiac myocytes, and elevation of either of these isoenzymes is also highly specific for acute coronary syndrome.1,2
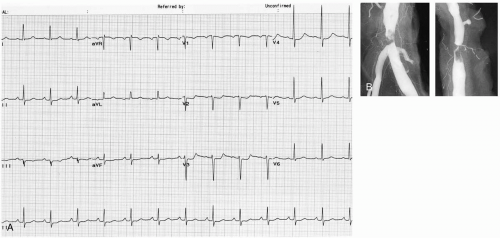 FIGURE 7-2 A: Sinus tachycardia (ST) segment depression in V2-V4, consistent with ischemia. (From Fowler, with permission.) B: Angiogram shows characteristic eccentric ragged stenosis with a small amount of attached intraluminal thrombus. (From Davies, with permission.) |
Clinical Complications
Patients with unstable angina are at increased risk for a myocardial infarction (MI) or sudden cardiac death.1
Management
Patients with unstable angina should be admitted to the hospital and given supplemental oxygen, aspirin (which reduces mortality and morbidity by 50% to 70%), nitrates, and β-blockers as well as antithrombotic treatment with unfractionated heparin or low-molecular-weight heparin (LMWH).1,2 Clopidogrel may further reduce morbidity and mortality when added to aspirin.1 The glycoprotein IIb/IIIa inhibitors may decrease the risks of recurrent angina and MI when used to treat unstable angina.1
REFERENCES
1. Cannon CP, Turpie AG. Unstable angina and non-ST-elevation myocardial infarction: initial antithrombotic therapy and early invasive strategy. Circulation 2003;107:2640-2645.
2. Parchure N, Brecker SJ. Management of acute coronary syndromes. Curr Opin Crit Care 2002;8:230-235.
7-3 Ventricular Aneurysm
Todd McGrath
Clinical Presentation
Ventricular aneurysms (VA) develop progressively over a 4- to 6-week period after infarction.1 Patients may have signs of left-sided congestive heart failure (CHF) (including pulmonary edema, third heart sound [S3] gallop, and jugular venous distention), thromboembolic phenomena secondary to mural thrombus formation, or ventricular arrhythmias.1,3 VAs may be detected on electrocardiographs (ECGs) of asymptomatic patients (sinus tachycardia [ST] elevations in the area of the aneurysm).
Pathophysiology
VA is a complication of a myocardial infarction (MI) that results in gradual thinning of the ventricular wall in the area of infarction. The thin-walled area may paradoxically bulge outward during ventricular contraction.1,2
Diagnosis
The diagnosis of VA should be suspected based on the clinical history in a patient with a recent AMI (within 4 to 6 weeks); it is confirmed by echocardiography.1
Clinical Complications
Clinical complications of VAs are related to the paradoxic outward bulging of the ventricle during systole and loss of uniform ventricular contraction.1 The patient may develop CHF secondary to poor left ventricular (LV) function. The aneurysm itself allows for the development of a thrombus within the ventricle that may cause thromboembolic phenomena. Finally, there is an increased risk for various potentially lethal ventricular arrhythmias.1,2 Rupture of a VA is an uncommon event.2
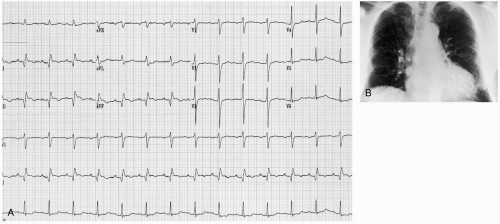 FIGURE 7-3 A: Persistent sinus tachycardia (ST) elevation consistent with ventricular aneurysm. (From Fowler, with permission.) B: Posterior chest radiograph of a patient with an aneurysm of the anterolateral wall of the left ventricle, which is enlarged. Note also the deformity of the left lower border of the cardiac silhouette. (From Kassner, with permission.) |
Management
Prevention is the key to management early in the postinfarction period. Control of heart rate, contractility, and afterload reduction help in the prevention of VA and in the initial medical treatment of an aneurysm. Thrombolytic therapy at the time of infarction has also been shown to reduce the incidence of VA.1 Patients presenting with VAs should receive immediate treatment of their presenting pathology (CHF, arrhythmias, embolic phenomena) and stabilization of airway, breathing, and circulation (ABCs) until definitive surgical correction of the aneurysm can be accomplished.1,2
REFERENCES
1. Bartel T, Vanheiden H, Schaar J, Mertzkirch W, Erbel R. Biomechanical modeling of hemodynamic factors determining bulging of ventricular aneurysms. Ann Thorac Surg 2002;74:1581-1588.
2. Das AK, Wilson GM, Furnary AP. Coincidence of true and false left ventricular aneurysm. Ann Thorac Surg 1997;64:831-834.
3. Tikiz H, Atak R, Balbay Y, Genc Y, Kutuk E. Left ventricular aneurysm formation after anterior myocardial infarction: clinical and angiographic determinants in 809 patients. Int J Cardiol 2002;82:7-16.
7-4 Pericarditis
Lisa Freeman
Clinical Presentation
Patients most commonly present to the emergency department (ED) with chest pain that may be either sharp and pleuritic, or dull and pressure-like. The pain radiates to the back, left shoulder, neck, arm, or trapezial ridge and is classically worse when supine and relieved by sitting up and leaning forward. Cough, dyspnea, dysphagia, nausea, and fever may be present. The cardinal sign of pericarditis on examination is the pericardial friction rub. The rub is often intermittent, positional, and best heard at the lower left sternal border.1
Pathophysiology
Pericarditis refers to inflammation and infection or infiltration of the pericardium, with or without pericardial effusion. It has many infectious and noninfectious causes.1
Inflammation of the pericardium usually is secondary to disorders in or about the heart, but in some cases it is caused by systemic disorders or by metastatic disease. Whatever the cause of the pericarditis, there is an inflammatory reaction of the epicardial and pericardial surfaces. The most frequent type of pericarditis, which is not bacterial in origin, produces serous fluid mixed with a fibrinous exudate. Bacterial pericarditis leads to a collection of purulent effusion in the pericardial sac. The most important causes of pericarditis are idiopathic (most common), viral, bacterial, tuberculous, fungal, parasitic, and neoplastic causes, as well as uremia, postmyocardial infarction (MI) complications, connective tissue diseases, radiation, chest trauma, and drugs such as hydralazine and procainamide.1
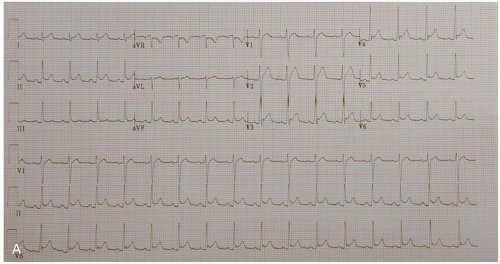 FIGURE 7-4 A: Electrocardiogram showing PR depression and diffuse sinus tachycardia (ST) elevation, as commonly seen in pericarditis. (Courtesy of Robert Hendrickson, MD.) |
Diagnosis
Diagnosis can be made with electrocardiograph (ECG) analysis. ECG changes occur in stages, and presenting patients may demonstrate any of the following. The first changes demonstrate diffuse sinus tachycardia (ST) elevation except in leads AVR and V1. The elevation is concave upward, and there are no reciprocal ST-T changes in other leads. This progresses to normalization of the ST segments and flattening of the T waves. The PR segment
may become depressed. The T waves may then become inverted. Eventually, all the abnormal changes resolve and the ECG may return to baseline. Laboratory evaluation may reveal leukocytosis and an elevated erythrocyte sedimentation rate (ESR). The chest radiograph is usually normal but may show cardiomegaly if there is a sizable pericardial effusion. An echocardiogram can exclude pericardial effusion. Pericardiocentesis may be performed to examine any pericardial fluid that is present, especially if a bacterial infectious cause is suspected or tamponade is present.1
may become depressed. The T waves may then become inverted. Eventually, all the abnormal changes resolve and the ECG may return to baseline. Laboratory evaluation may reveal leukocytosis and an elevated erythrocyte sedimentation rate (ESR). The chest radiograph is usually normal but may show cardiomegaly if there is a sizable pericardial effusion. An echocardiogram can exclude pericardial effusion. Pericardiocentesis may be performed to examine any pericardial fluid that is present, especially if a bacterial infectious cause is suspected or tamponade is present.1
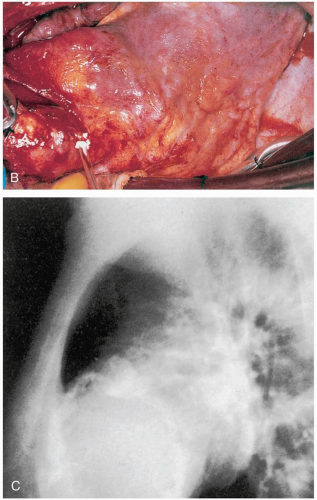 FIGURE 7-4 cont’d. B: Operative view of the heart showing the epicardial surface with a fine granular appearance. (From Hurst, with permission.) C: Lateral chest radiograph showing calcified pericardium surrounding the heart. (From Hurst, with permission.) |
Clinical Complications
Pericarditis usually results in at least a small amount of excess pericardial fluid. In some cases, the fluid accumulation becomes large and leads to cardiac tamponade. Constrictive pericarditis may develop secondary to chronic inflammation, resulting in thickening and adherence of the pericardium to the heart.1
Management
Pericarditis without significant pericardial effusion needs no specific treatment other than pain control. Most patients can be discharged from the ED with antiinflammatory medication and follow-up within 48 hours. These patients should also be instructed to return to the ED if symptoms worsen. Pericarditis that may be secondary to a systemic illness usually requires hospital admission for diagnosis and management of the primary condition.1
REFERENCES
1. Spodick DH. Acute pericarditis: current concepts and practice. JAMA 2003;289:1150-1153.
7-5 Pericardial Effusion and Tamponade
Robert Chisholm
Clinical Presentation
The classic manifestation of acute pericardial tamponade is Beck’s triad: hypotension, jugular venous distention, and muffled heart sounds. It may present insidiously, with a slowly developing effusion not exhibiting this triad. These patients may present with dyspnea and exercise intolerance.1
Pathophysiology
The most common cause of pericardial effusion with tamponade is malignancy. Other important causes are tuberculosis, uremia, hemorrhage (excessive anticoagulation), myxedema, systemic lupus erythematosus (SLE), radiation therapy, and chronic pericarditis.1
A pericardial effusion involves accumulation of fluid in the potential space between the fibrinous outer pericardium and the serous inner pericardium. This space normally contains about 20 mL of plasma-like fluid. Hemodynamic compromise can occur with an acute (minutes to hours) increase in a small amount of fluid (as little as 50 mL) or with a more lengthy (weeks to months) accumulation of fluid (up to 120 mL). However, if the intrapericardial pressure matches or exceeds ventricular diastolic pressures, systemic venous congestion occurs, cardiac output falls, and hypotension results.1
Diagnosis
Diagnosis is suggested by history and clinical examination. Examination may show the clinical findings of tamponade, such as jugulovenous distention, hypotension, distant heart sounds, pulsus paradoxus, and narrow pulse pressure. Chest radiography may show an enlarged heart if the effusion is chronic, and the electrocardiograph (ECG) may have low-voltage QRS complexes or electrical alternans suggestive of effusion. Emergency echocardiography should be performed for all suspected cases of pericardial tamponade. Ultrasonography shows fluid in the pericardial space and impaired cardiac motion. Inferior vena cava dilatation without inspiratory collapse is also seen and is highly suggestive of tamponade. If echocardiography is not immediately available but the patient is in critical condition and suspicion for tamponade exists, pericardiocentesis may be both diagnostic and therapeutic. Definitive intervention by a general or cardiothoracic surgeon is required.1
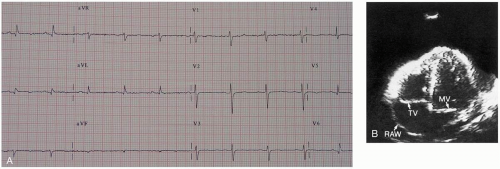 FIGURE 7-5 A: Electrocardiogram showing electrical alternans as seen in pericardial effusion. (Courtesy of John Fojtik, MD.) B: Apical four-chamber echocardiogram showing right atrial wall (RAW) collapsing inwardly, suggesting early tamponade. TV, tricuspid valve; MV, mitral valve. (From Hurst, with permission.) |
Clinical Complications
Pericardial tamponade, if not identified and treated, can lead to impaired cardiac output, hypotension, and death.1
Management
Patients with pericardial tamponade and hemodynamic instability should have immediate pericardiocentesis, followed by definitive surgical intervention and admission to the intensive care unit (ICU). Patients without hemodynamic instability should be treated with intravenous (IV) fluids, and urgent consultation with a thoracic surgeon should be obtained. Those with identified cardiac effusions with no tamponade physiology should be admitted to the cardiology inpatient unit or equivalent for telemetry monitoring and further evaluation. Pericardial effusions and tamponade in a nontrauma patient are suspicious for malignancy, and further workup is needed.1
REFERENCES
1. Spodick DH. Pathophysiology of cardiac tamponade. Chest 1998;113:1372-1378.
7-6 Congestive Heart Failure
Robert Chisholm
Clinical Presentation
Patients with congestive heart failure (CHF) typically complain of shortness of breath and dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, a cough productive of pink frothy sputum, weakness, lightheadedness, abdominal pain, malaise, and nausea.
Pathophysiology
CHF may develop from a variety of causes, including valvular disease, ischemic cardiomyopathy (CM), dysrhythmia, hypertensive cardiomyopathy (HCM), constrictive heart disease, and dilated cardiomyopathy (DCM).2 The end result is an imbalance in the myocardial Starling forces, from either inadequate filling or inadequate emptying.1,2
Diagnosis
The diagnosis is typically made clinically and confirmed with chest radiography, echocardiography, β-natriuretic peptide (BNP), or some kind of these. Examination may reveal tachypnea, jugular venous distention, peripheral edema, hypertension, pulmonary rales and rhonchi, a laterally displaced cardiac apical impulse, and S3 or S4 heart sounds. Chest radiography may reveal cardiomegaly; an increase in the prominence of the pulmonary vasculature, particularly in the apical areas (“cephalization”); and Kerley’s lines. Electrocardiograph (ECG) may be used to determine the presence of myocardial ischemia. Laboratory analysis may reveal hepatic congestion or renal hypoperfusion. BNP is released from myocardial cells with stretching and is often elevated in CHF. BNP may be particularly helpful as a diagnostic test if the cause of dyspnea is in doubt (e.g., chronic obstructive pulmonary disease [COPD] versus CHF).
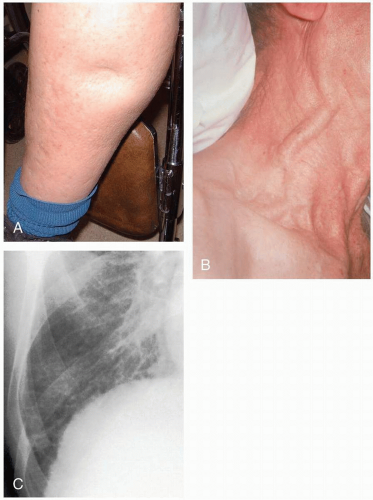 FIGURE 7-6 A: Pitting edema. (Courtesy of Mark Silverberg, MD.) B: Neck vein distention. (Courtesy of Michael Greenberg, MD.) C: Chest radiograph demonstrating pulmonary interstitial edema with Kerley’s B lines. (From Hurst, with permission.) |
Clinical Complications
Complications may include pulmonary edema, respiratory failure, end-organ hypoperfusion, shock, and death.
Management
Patients in extremis may require high-flow oxygen, large-bore intravenous (IV) access, endotracheal intubation, and inotropic support. In patients with mild to moderate symptoms, a determination of the cause of the heart failure is an initial priority, because it leads therapy. Initial steps may include administration of oxygen and elevation of the head of the bed.2 Typical therapeutic strategies aim to decrease preload (nitrates, angiotensin-converting enzyme [ACE] inhibitors, diuretics, BNP), decrease afterload (ACE inhibitors, nitroprusside, BNP), increase inotropic support (dobutamine, dopamine), and decrease anxiety and myocardial oxygen demand (opioids).1,2
Patients with new-onset CHF should be admitted to the hospital on a telemetry ward for further evaluation. Patients with chronic heart failure with a clear inciting cause (e.g., increased water intake) and no suspicion of an acute event (e.g., myocardial infarction [MI], dissection, pulmonary embolism [PE]) who improve in the emergency department (ED) may be discharged to home with close follow-up.1,2
REFERENCES
1. Jessup M, Brozena S. Heart failure. N Engl J Med 2003;348:2007-2018.
2. Millane T, Jackson G, Gibbs CR, Lip GY. ABC of heart failure: acute and chronic management strategies. BMJ 2000;320:559-562.
7-7 Dilated Cardiomyopathy
Lisa Freeman
Clinical Presentation
Patients may present with symptoms of left-sided congestive heart failure (CHF) that have developed gradually. Shortness of breath, chest pain, fatigue, and weakness may be present. Physical examination often reveals S3 and S4 murmurs that reflect atrioventricular (AV) valve regurgitation, pulmonary rales, and signs of systemic shock. The blood pressure may be normal or low, and there is often tachycardia if CHF has developed. If right-sided heart failure develops, patients have jugular venous distention, ascites, and peripheral edema. Some patients have minimal or no symptoms, whereas others have progressive deterioration leading to death in 1 to 5 years.1
Pathophysiology
Dilated cardiomyopathy (DCM) refers to cardiac enlargement and impaired contraction of the left ventricular (LV) or of both ventricles. The causes are variable and include cardiovascular disease, alcohol- or drug-related, familial, viral, immune, and idiopathic causes. The estimated incidence is 5 to 8 cases per 100,000 population per year and is increasing.1
DCM is the most common form of cardiomyopathy (CM), accounting for 60% of all cases. It is characterized by ventricular dilatation and contractile dysfunction. The coronary arteries are often normal, and chest pain in these patients is thought to be caused by subendocardial ischemia. Excessive ethanol intake is a major cause of DCM, accounting for one third of cases. Ethanol may have a direct toxic effect on the myocardial cells, or secondary nutritional effects (e.g., lack of thiamine) in alcoholic patients may be contributory. Genetics is also thought to play a factor, because 20% of patients have a first-degree relative with DCM.1
Diagnosis
Identification of the reversible causes of DCM (ethanol, cobalt, mercury, lead, antiretroviral drugs, thiamine deficiency, hypothyroidism, acromegaly, thyrotoxicosis, hypocalcemia, hypophosphatemia, toxoplasmosis, and sarcoidosis) is particularly important during the first presentation.1
The goal of emergency department (ED) care is to exclude potentially reversible causes in a patient who has a clinical presentation suspicious for DCM. These include hypophosphatemia, hypocalcemia, ethanol, mercury, lead, antiretroviral agents, thiamine deficiency, toxoplasmosis, sarcoidosis and hyperthyroidism, and hypothyroidism.1 Chest radiography usually reveals generalized cardiomegaly and pulmonary vascular redistribution. Interstitial and alveolar pulmonary edema are less common on initial presentation. Electrocardiograph (ECG) reveals sinus tachycardia when heart failure is present. Poor R-wave progression is seen, as is an interventricular conduction delay. Virtually any dysrhythmia may be seen, although atrial fibrillation (AF) is common, especially in patients with alcohol-induced DCM. Echocardiography makes the definitive diagnosis. It also is useful to assess the degree of LV dysfunction and to exclude valvular or pericardial disease. Myocardial biopsy may be needed if a reversible cause is suspected.
Clinical Complications
Intracavitary thrombi are common and may lead to systemic emboli. Pulmonary embolism (PE) is also a known complication. Prominent causes of death in patients with DCM include fulminant heart failure and dysrhythmias.
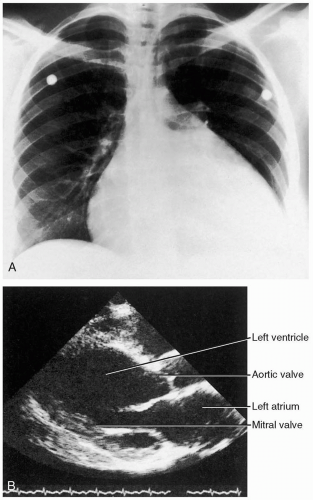 FIGURE 7-7 A: Chest radiograph in a patient with cardiomyopathy, demonstrating cardiomegaly with biventricular enlargement and pulmonary venous hypertension. (From Kassner, with permission.) B: Cross-sectional echocardiogram in the long-axis view recorded during systole in a patient with dilated cardiomyopathy. The systolic dimension of the left ventricle was 7.4 cm, and the diastolic dimension was 8.6 cm. (From Becker, with permission.) |
Management
The basic management for idiopathic DCM is the same for CHF. Useful therapeutic agents include vasodilators (angiotensin-converting enzyme [ACE] inhibitors, nitrites, hydralazine), diuretics, digoxin, and carvedilol. Patients with DCM due to alcohol abuse will benefit from abstaining from alcohol, because the early manifestations may reverse.
REFERENCES
1. Dec GW, Fuster V. Medical progress: idiopathic dilated cardiomyopathy. N Engl J Med 1994;331:1564-1575.
7-8 Hypertrophic Cardiomyopathy
Robert Chisholm
Clinical Presentation
Hypertrophic cardiomyopathy (HCM) is the most common genetic cardiovascular disease (0.2% of the population). Patients may present with a variety of problems, including dyspnea, syncope, presyncope, angina, palpitations, congestive heart failure (CHF), and sudden death.1,2 Patients may develop sudden cardiac death from ventricular outflow obstruction or arrhythmias. Patients are typically young and previously asymptomatic, and sudden cardiac death may occur after physical exertion.
Examination may reveal cardiac arrhythmias, signs of CHF, a systolic ejection crescendo-decrescendo murmur that is heard best between the apex and left sternal border (and increases with Valsalva maneuvers), and the holosystolic murmur of mitral regurgitation.1
There are no specific laboratory studies. The electrocardiograph (ECG) is abnormal in 90% of people with HCM, but there are no abnormalities that are specific for the disease. Chest radiography may be normal or show cardiomegaly. In severe HCM, radiographic findings of CHF may be present. Patients who are considered to be at risk for adverse events related to HCM should undergo Holter monitoring for surveillance of ventricular tachycardia and exercise testing to gauge the presence and severity of exercise-induced hypotension.1
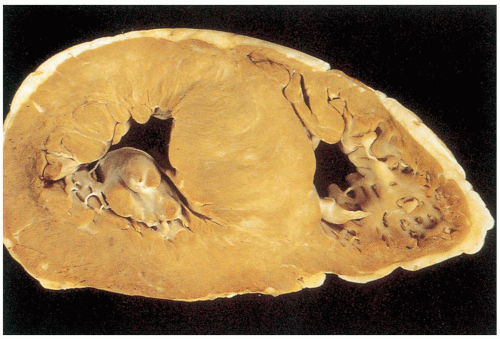 FIGURE 7-8 Cross-section through a heart with hypertropic cardiomyopathy and asymmetry of the affected interventricular septum. (From Becker, with permission.) |
Pathophysiology
HCM is a genetic disorder that is characterized by the inappropriate, asymmetric hypertrophy of the left ventricular (LV).1
Patients with HCM may have asymmetric septal hypertrophy. In addition to obstruction from the hypertrophy, patients may have further obstruction of the left ventricular outflow tract from systolic anterior motion of the mitral valve. Greater ventricular contractile force leads to higher ejection velocity, which translates to increased displacement of the mitral valve, resulting in further narrowing of the outflow tract.1
Diagnosis
The ECG is abnormal in 90% of patients, showing left ventricular hypertrophy (LVH) with Q waves in the anterior and lateral leads.1 Echocardiography is the diagnostic study of choice for suspected HCM. The diagnosis is suggested by the finding of a hypertrophied but nondilated left ventricle in the absence of illness capable of producing the hypertrophy.1
Clinical Complications
Management
Patients presenting to the emergency department (ED) with new-onset heart failure, chest pain, or syncope should be admitted for further workup. β-blockers may be used for heart failure or AF, because they decrease heart rate, increase diastolic filling time, and decrease the LV outflow tract obstruction.2 Any child or young person who presents with cardiac arrhythmia or syncopal/presyncopal complaints may be at risk for sudden cardiac death. If the diagnosis is suggested in a child or adult with new-onset symptomology, an echocardiogram and cardiology consultation are warranted.2
REFERENCES
1. Maron BJ. Hypertrophic cardiomyopathy: a systemic review. JAMA 2002;287:1308-1320.
2. Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997;336: 775-785.
7-9 High-Output Cardiac Failure
Jennifer Larson
Clinical Presentation
The characteristic physical findings are all related to the hyperdynamic state and include relative tachycardia, bounding pulses with a quick upstroke, wide pulse pressure, venous hum (usually over deep internal jugular veins and occasionally over the femoral veins), systolic bruit over the carotids, S3 gallop, and midsystolic murmur. Patients with chronic high-output heart failure typically present with signs and symptoms of low-output (or “congestive”) heart failure, with systemic congestion and pulmonary edema. Acute presentations correlate with the underlying cause. Some specific causes have obvious physical findings, such as congenital fistulas (cutaneous hemangiomas or hemorrhagic telangiectasia) or hyperthyroidism (exophthalmos and a fine tremor). Laboratory data and radiologic studies are directed toward suspected causes. Chest radiography confirms pulmonary edema if there is associated congestive heart failure (CHF) in the chronic setting. If the cause is unknown, evaluation of hemoglobin/hematocrit, thyroid studies, thiamine concentration, aminotransferase levels, and a coagulation profile may be prudent.
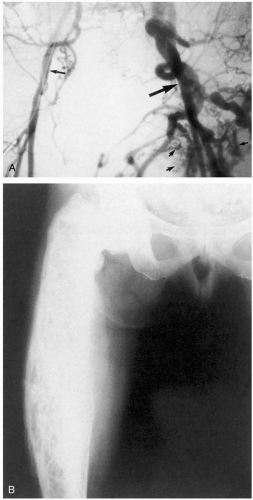 FIGURE 7-9 A: Arteriovenous communication. (From Hurst, with permission.) B: Paget’s disease. (From Kassner, with permission.) |
Pathophysiology
High-output cardiac failure is defined as heart failure in the presence of elevated cardiac output and low systemic vascular resistance.1
Several physiologic conditions may cause an increase in cardiac output (pregnancy, fever, exercise); however, heart failure usually does not occur in these cases unless there is an underlying cardiac pathology. Disorders associated with the development of high-output cardiac failure include hyperthyroidism, anemia, “wet” beriberi (thiamine deficiency), renal disease, hepatic disease, systemic arteriovenous fistulas, polycythemia vera, and carcinoid syndrome.
Diagnosis
Diagnosis depends on the primary condition.
Clinical Complications
Complications include acute coronary ischemia from high cardiac output, typically seen with underlying atherosclerotic disease, and CHF from the chronic high-output state.
Management
Management is best aimed at treatment of the underlying cause (e.g., hyperthyroidism, anemia, atrioventricular (AV) fistula, hemangioma, Paget’s disease). If the patient is clinically stable in the emergency department (ED), close follow-up with outpatient workup and treatment of the primary condition is acceptable. The patient may require admission for treatment of CHF and, in the setting of low systemic vascular resistance, may require vasopressor therapy.
REFERENCES
1. Braverman AC, Steiner MA, Picus D, White H. High-output congestive heart failure following transjugular intrahepatic portal-systemic shunting. Chest 1995;107:1467-1469.
7-10 Abdominal Aortic Aneurysm
Lisa Freeman
Clinical Presentation
The majority of abdominal aortic aneurysms (AAA) are asymptomatic, and patients who present to the emergency department (ED) usually do so when the AAA expands or is leaking (bleeding). The pain is often nonspecific in location and character, and it is necessary to have a high index of suspicion to make a diagnosis. AAAs may also mimic other conditions that cause trunk pain, such as renal colic and mechanical low back pain. A ruptured AAA can cause signs of rapid blood loss, including syncope, hypotension, and tachycardia, as well as a pulsatile abdominal mass and abdominal tenderness.2,3,4
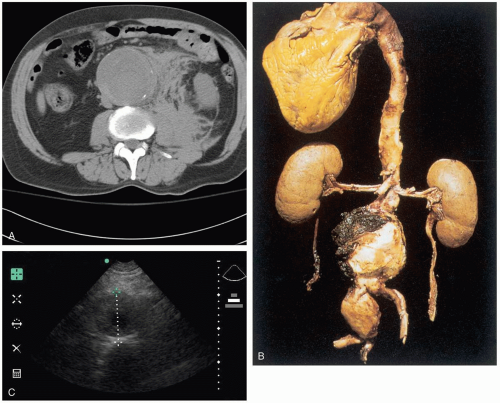 FIGURE 7-10 A: An 8.3 cm infrarenal abdominal aortic aneurysm (AAA) with peritoneal blood. (Courtesy of Robert Hendrickson, MD.) B: Artherosclerotic AAA located distal to the renal arteries. There is an additional aneurysm in the right common iliac artery. The atherosclerosis of the aorta is associated with obstructive atherosclerotic coronary artery disease. (From Hurst, with permission.) C: Transverse view of infrarenal AAA. (Courtesy of Al Sabbaj, MD.) |
Pathophysiology
An AAA is defined as a focal dilatation of the abdominal aorta that results in a diameter at least 50% larger than the expected normal diameter.1 The exact diameter of a normal aorta is variable, depending on gender, body habitus, and age, but typically ranges from 17 to 24 mm. A diameter of 30 mm is generally accepted as aneurysmal dilatation. AAAs are rare before the age of 50 years, but they are found in 2% to 5% of patients older than 65 years of age. At least 15,000 patients die from ruptured AAAs each year in the United States.1
Several biochemical aberrations can occur that lead to the loss of elastin and collagen in the aortic wall. These
structural changes result in degeneration and aneurysmal dilatation. The risk of rupture increases with aneurysm size and significantly increases after the aneurysm reaches 5 cm or greater. Intraperitoneal rupture usually is rapidly fatal. Retroperitoneal rupture may be contained, giving the patient time to present to the ED for treatment. Risk factors for AAA include advanced age, gender (males carry higher risk), hypertension, and smoking. Conditions such as chronic obstructive pulmonary disease (COPD), coronary artery disease, peripheral vascular disease (PVD), or a predisposed family history also contribute to the risk of developing AAA.1,2,3,4
structural changes result in degeneration and aneurysmal dilatation. The risk of rupture increases with aneurysm size and significantly increases after the aneurysm reaches 5 cm or greater. Intraperitoneal rupture usually is rapidly fatal. Retroperitoneal rupture may be contained, giving the patient time to present to the ED for treatment. Risk factors for AAA include advanced age, gender (males carry higher risk), hypertension, and smoking. Conditions such as chronic obstructive pulmonary disease (COPD), coronary artery disease, peripheral vascular disease (PVD), or a predisposed family history also contribute to the risk of developing AAA.1,2,3,4
Diagnosis
The diagnostic approach taken depends on the stability of the patient and the index of suspicion. An unstable patient with a convincing presentation should receive immediate attention to their airway, breathing, and circulation (ABCs), followed by intravenous (IV) access and emergency surgical consultation. Blood products should also be available. Surgical consultation should not be delayed for any diagnostic study, because the only treatment of a ruptured AAA is immediate surgery.2,3,4 Palpation of a pulsatile mass in the epigastrium should be attempted. However, palpation cannot be used to rule out a diagnosis of AAA, because the physical examination is only 68% sensitive in detecting an enlarged aorta (up to 82% sensitive for AAAs larger than 5 cm).3
Ultrasonography can be done at the bedside to attempt visualization of an enlarged aorta for rapid confirmation of the diagnosis. Bedside ultrasound should not be used to identify peritoneal blood (focused abdominal sonography for trauma [FAST] examination), because most bleeding is retroperitoneal, and the lack of peritoneal blood cannot be used to rule out AAA. Stable patients should undergo computed tomography (CT) with IV contrast, which is almost 100% sensitive in detecting AAA.2 All patients with a symptomatic AAA should have a complete blood count, typing and cross-matching for 6 units of packed red blood cells (PRBCs), coagulation studies, and creatinine administration.2,3,4
Clinical Complications
Management
Patients should have immediate assessment of their ABCs, placement of two large-bore IV access tubes, use of a cardiac monitor, and administration of oxygen. Patients with significant shock require IV fluids and unmatched blood products. Ruptured AAA requires immediate surgical intervention. Intubation may also be required.1,2,3,4
REFERENCES
1. Shames ML, Thompson RW. Abdominal aortic aneurysms: surgical treatment. Cardiol Clin 2002;20:563-578.
2. Clinical policy: critical issues for the initial evaluation and management of patients presenting with a chief complaint of nontraumatic acute abdominal pain. Ann Emerg Med 2000;36:406-415.
3. Fink HA, Lederle FA, Roth CS, Bowles CA, Nelson DB, Haas MA. The accuracy of physical examination to detect abdominal aortic aneurysm. Arch Intern Med 2000;160:833-836.
4. Lederle FA, Simel DL. The rational clinical examination: does this patient have abdominal aortic aneurysm? JAMA 1999;281:77-82.
7-11 Aortic Dissection
Samuel Kim
Clinical Presentation
Patients with aortic dissection commonly present to the emergency department (ED) with complaints of chest pain or back pain or both. The pain is usually abrupt in onset (85%) and is classically described as a “tearing” or “ripping” pain (51%). The location of the pain is most commonly the chest (73%), followed by the back (53%) and then the abdomen (30%).2 Depending on the extent of the dissection, perfusion of branch arteries of the aorta may be affected, resulting in a myriad of possible ischemic presentations. Neurologic findings occur in 18% to 30% of cases, caused by stroke/cerebral ischemia, decreased spinal cord perfusion, or compression of peripheral nerves. Renal ischemia with resultant renal failure may occur. Mesenteric artery involvement may also cause mesenteric ischemia with acute abdominal pain. Pulse pressure and blood pressure differentials between the upper extremities may be seen (38%) and are the most specific physical examination finding. Lower-extremity perfusion may also be compromised, with symptomatic ischemia seen in 15% to 20% of cases. Involvement of the ascending aorta may cause compromise of coronary artery perfusion with resultant ischemia. Acute aortic valve insufficiency and pericardial effusion with tamponade may occur, with resultant hypotension and shock. The electrocardiograph (ECG) is often nonspecific and varies depending on coronary artery involvement. Chest radiography shows mediastinal widening in up to 50% of cases but is normal in up to 12% of cases.2 Other findings may include changes in the position of the aorta, double aortic shadow, enlargement of the aortic knob, displacement of aortic calcifications, left pleural effusion, and disparity in the sizes of the ascending and descending aorta.
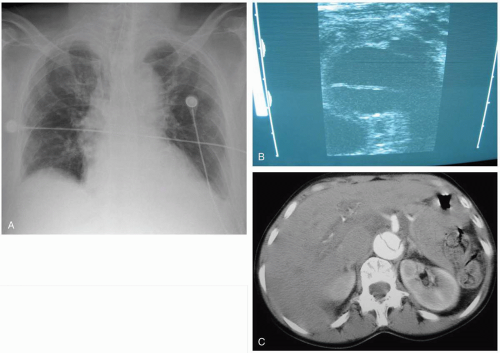 FIGURE 7-11 A: Chest radiograph showing widened mediastinum and displacement of trachea and esophagus. (Courtesy of Robert Hendrickson, MD.) B: Ultrasonogram showing a false lumen filled with clot. (Courtesy of Mark Silverberg, MD.) C: Computed tomogram showing aortic dissection with false lumen. (Courtesy of Colleen Campbell, MD.) |
Pathophysiology
Aortic dissection refers to separation of the layers of the aortic wall, usually caused by a tear in the intima. Aortic dissections are classified by two different schematics, the Stanford and DeBakey systems. Under the Stanford classification, dissections involving the ascending aorta are type A, and type B dissections involve only the descending aorta. Under the DeBakey classification, type I dissections include the ascending aorta, aortic arch, and descending aorta, whereas type II dissections involve only the ascending aorta and type III involve only the descending aorta.1
Aortic dissection may result from an initial intimal tear followed by cleavage of the intimal and medial layers and subsequent extension. Much more rarely, cleavage of the intimal and medial layers is caused by an initial intramural hematoma that expands and then ruptures through the intima. Usually, the intima tears around areas with the greatest pressure gradients and fluctuations, most commonly in the ascending aorta and the first portion of the descending aorta.2 The ratio of type A to type B dissections is approximately 2:1. There is a male-to-female ratio of approximately 2:1, and the peak incidence is in the sixth and seventh decades of life.3 Other risk factors include long-standing untreated hypertension, aortic dilatation, aneurysm, coarctation, aortic arteritis, aortic arch hypoplasia, chromosomal aberrations (Turner’s and Noonan’s syndromes), and connective tissue diseases (Marfan’s and Ehlers-Danlos syndromes). Iatrogenic trauma has also been implicated. Cocaine users and pregnant women have been cited as higher-risk population groups.3
Diagnosis
All patients in whom aortic dissection is suspected should be imaged with transesophageal echocardiography (TEE), magnetic resonance imaging (MRI), computed tomography (CT), or angiography. Any modality except transthoracic echocardiography (TTE) may be used to rule out aortic dissection if the clinical suspicion is low. If the pretest probability is moderate to high, a second test should be ordered if the first test is negative.4 The selection of the second test should be made with time delays in mind, because time to diagnosis and treatment significantly affects survival. TEE and MRI are the most sensitive and specific studies, with TEE having a sensitivity of 95% to 100% and a specificity of 70% to 95%, and MRI having a sensitivity of 95% and a specificity of 100%. CT has a sensitivity of 83% to 90% and a specificity of 90% to 100%.4
Clinical Complications
Aortic dissection may result in rupture, with type A dissections being most prone to rupture. The mortality rate after rupture is 1% per hour after onset of symptoms.4 Aortic dissection may also result in stroke, shock, hypotension, acute renal failure, neurologic deficits, symptomatic limb ischemia, and mesenteric ischemia.
Management
Patients with aortic dissection should receive rapid control of hypertension and tachycardia with intravenous (IV) antihypertensive medications, regardless of dissection type. A combination of a β-blocker and a vasodilator (e.g., nitroprusside) usually is used,3 although single therapy with labetalol or esmolol also may be used. Patients with probable aortic dissection should have immediate consultation with an appropriate surgeon or cardiologist or both. Definitive treatment is based on the type of dissection. Type A dissections are treated by surgery unless comorbid conditions preclude surgery as an option, and type B dissections are managed medically. All aortic dissections require intensive care unit (ICU) admission.1,2,3,4
REFERENCES
1. Pretre R, Von Segesser L. Aortic dissection. Lancet 1997;349:1461-1464.
2. Hagan PG, Nienaber CA, Isselbacher EM, et al. The International Registry of Acute Aortic Dissection (IRAD): new insights into an old disease. JAMA 2000;283:897-903.
3. Sarasin FP, Louis-Simonet M, Gaspoz JM, Junod A. Detecting acute thoracic aortic dissection in the emergency department: time constraints and choice of the optimal diagnostic test. Ann Emerg Med 1996;28:278-288.
4. Khan I, Nair C. Clinical, diagnostic, and management perspectives of aortic dissection. Chest 2002;122:311-328.
7-12 Thoracic Aortic Aneurysm
Robert Hendrickson
Clinical Presentation
Patients with a rupture of a thoracic aortic aneurysm (TAA) typically develop rapid exsanguination and rarely survive to hospitalization. If they survive to the emergency department (ED), these patients have hypovolemic shock and evidence of exsanguination (widened mediastinum and hemothorax) on chest radiography. Occasionally, patients present with chest pain that is attributable to rapid expansion of the aneurysm and impending rupture.1
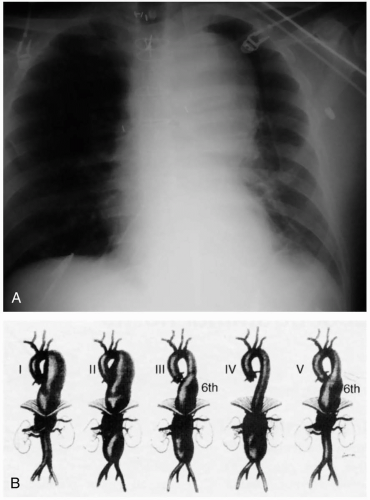 FIGURE 7-12 A: Thoracic aneurysm. (Courtesy of Donald Sallee, MD.) B: Thoracoabdominal aortic aneurysm classification: I, between left subclavian and renal arteries; II, between left subclavian and bifurcation; III, between 6th rib and bifurcation; IV, between the 12th rib and the bifurcation; V, between the 6th rib and the renal arteries. (From Safi HJ, Winnerkvist A, Miller CC 3rd, et al. Effect of extended cross-clamp time during thoracoabdominal aneurysm repair. Ann Thorac Surg 1998;66:1204-1209, with permission.) |
Patients with unruptured TAAs may present with symptoms that are related to compression of thoracic structures, such as hoarseness, dysphagia, dyspnea, chest pain, or superior vena cava syndrome.2 TAA may also be detected on routine chest radiography as a widened inferior mediastinum and enlarged aortic shadow. An enlarged aorta may be evident on abdominal examination if the aneurysm extends to the abdomen.
Pathophysiology
TAA refers to aneurysmal dilatation of the ascending (50%), arch (10%), or descending (40%) portion of the aorta within the thorax.1 Aneurysms may be primarily thoracic or thoracoabdominal.1
Aneurysmal dilatation may be related to atherosclerosis, trauma, medial degeneration (Marfan syndrome), aortitis (syphilis), mycotic infections, and vasculitis (Takayasu’s arteritis).1,2 Dilatation of the proximal aorta may also occur with bicuspid valves and after surgical repair of aortic dissection.2
Diagnosis
Diagnosis of an unruptured TAA typically is made on chest radiography and should be confirmed with a thoracic computed tomography (CT) to differentiate aneurysmal dilatation from anatomic tortuosity of the aorta.
Clinical Complications
Management
Patients with a ruptured TAA or impending rupture require oxygen, large-bore fluid and blood infusions, emergency surgical consultation, and rapid transport to the operating theater. Patients without rupture who have radiographic signs of aortic dilatation should be further evaluated with a thoracic CT and close follow-up with a thoracic surgeon. Control of hypertension and smoking cessation may also be helpful.2
REFERENCES
1. Fann JI. Descending thoracic and thoracoabdominal aortic aneurysms. Coron Artery Dis 2002;13:93-102.
2. Moon MR, Sundt TM 3rd. Aortic arch aneurysms. Coron Artery Dis 2002;13:85-92.
7-13 Septal Wall Rupture
Todd McGrath
Clinical Presentation
Patients with septal wall rupture (SWR) present with chest pain, shortness of breath, and signs of cardiogenic shock. This condition is typically seen either within the first 24 hours of an acute myocardial infarction (AMI) or 3 to 5 days after myocardial infarction (MI). Patients may have a new, harsh systolic murmur at the left sternal border.1
Related posts:







Full access? Get Clinical Tree


