Cardiogenic Shock Complicating Acute Coronary Syndromes
John M. Field
Patients with cardiogenic shock and STEMI should be primarily transported or secondarily transferred (with a door-to-departure time of within 30 minutes) to facilities capable of invasive strategies such as insertion of an intra-aortic balloon pump (IABP), percutaneous coronary intervention (PCI), and coronary artery bypass grafting (CABG). Recent trial data have shown an improved prognosis for survivors of cardiogenic shock who benefit from an early invasive strategy. Thus an aggressive approach to achieve early and complete reperfusion of the infarct related artery (IRA), to prevent or correct mechanical complications of MI, and to provide pharmacologic and mechanical interventions to favorably influence early infarct remodeling are warranted in patients with cardiogenic shock or at high-risk for this complication.
The incidence of cardiogenic shock had largely remained unchanged but appears to be decreasing in parallel with increasing rates of primary PCI for STEMI. And mortality is now, in the modern era, ∼50% below historical levels of 80% to 90%.
Right ventricular (RV) shock (usually found in association with inferior wall MI) has a high mortality rate, similar to that of left ventricular (LV) shock.
An invasive strategy is recommended for patients <75 years of age; carefully selected patients age ≥75 can also receive aggressive early revascularization. If heart failure and shock develop after hospital admission or if shock develops within the first 36 hours of onset of MI, the patient should undergo diagnostic angiography and PCI or CABG if possible.
This chapter describes complications of acute coronary syndromes (ACS): shock, pulmonary edema, and hypotension. The first section discusses a general approach to the patient presenting with undifferentiated hypotension (see also Chapter 8). The second section summarizes the pathophysiology and treatment of cardiogenic shock associated with ST-segment elevation myocardial infarction (STEMI) and other ACS, emphasizing therapy unique to these patients. The last section reviews the algorithm for acute pulmonary edema, hypotension, and shock and details the initial evaluation and stabilization of any patient with pulmonary edema and shock or hypertensive urgency. It also contains more information about treatment decisions based on the initial response to therapy.
Epidemiology
Cardiogenic shock remains the leading in-hospital cause of mortality. In general, cardiogenic shock is due to decreased systemic cardiac output in the presence of adequate intravascular volume, resulting in tissue ischemia and cellular hypoxia. In 75% of patients with STEMI, cardiogenic shock is most often caused by extensive MI. But while cardiogenic shock may be secondary to a large STEMI, it can also be due to NSTEMI and potentially reversible conditions such as mechanical complications of MI or multivessel disease with reversible ischemia in the non–infarct-related arteries.
Historically, during the advent of the reperfusion era, the incidence of cardiogenic shock remained unchanged at about 7.5% and prognosis was poor, with in-hospital mortality exceeding 77%.2 The incidence of cardiogenic shock in large trials of fibrinolysis is variable but has also ranged from 4% to 7%.3,4,5 Overall, the prognosis of patients with cardiogenic shock has largely remained unchanged. However, recent data indicate that survivors of cardiogenic shock may have a reasonable and improved prognosis. In a 6-year follow-up of the SHOCK trial,6 long-term survival was 62.4% in the revascularized group and 44.4% in the aggressively managed medical group. Thus, an aggressive approach—to achieve early and complete reperfusion of the IRA, prevent or correct mechanical complications of MI, and provide pharmacologic and mechanical interventions to
favorably influence early infarct remodeling—is warranted.7,8,9
favorably influence early infarct remodeling—is warranted.7,8,9
Hypotension and Shock
Hypotension is defined as a systolic blood pressure <90 mm Hg or an acute fall in blood pressure ≥30 mm Hg from baseline in previously hypertensive patients.1 In patients with MI, hypotension can be due to other causes in addition to impaired ventricular function. In considering the differential diagnosis of hypotension and shock even in patients with MI, secondary causes that may complicate management should be kept in mind. For example, these may include bleeding from procedures or anticoagulation, sepsis, and inadequate volume or preload conditions.
Shock is a clinical condition characterized by a sustained and significant reduction in blood flow and oxygen delivery to organs and tissues. It is important to realize that shock and low blood pressure, although related, are not the same. In basic terms, shock is a condition in which tissue oxygenation (and cellular ventilation and nutrition) is inadequate for demand.
Patients frequently present with shock and no immediately obvious etiology (i.e., undifferentiated shock). Blood pressure alone can be misleading or “normal” for a variety of reasons. Hence the diagnosis of shock is a clinical one characterized by several of the following findings:
Clinically ill appearance or altered mental status
Low blood pressure (defined as a systolic blood pressure [SBP] <80 to 90 mm Hg)
Tachycardia (heart rate >100)
Tachypnea (respiratory rate >22 breaths/min or PaCO2 <32 mm Hg)
Systemic acidosis (serum lactate >4 mmol/L)
Decreased urine output (<0.5 mL/kg/hr)
Differential Diagnosis of Shock
Not all of these criteria may be present. For example, patients on beta-blockers may not have tachycardia. In early shock, blood pressure may be normal or only slightly low because of excess adrenergic drive, and it may not drop significantly until late in the process.
The clinician can use provisional etiologic mechanisms to characterize shock and to identify the appropriate initial focus of therapy:
Arrhythmic shock → antiarrhythmic therapy
Hypovolemic shock → volume therapy
Cardiogenic shock → reperfusion for STEMI; support of ventricular function
Distributive shock → vasoactive drug therapy
An etiologic approach to shock often oversimplifies the problem. Any patient with severe or sustained shock will likely require some support of heart rate and rhythm, titration of fluid therapy to optimize intravascular volume, support of ventricular function, and manipulation of vascular resistance and distribution of blood flow. All patients with severe or sustained shock will have some myocardial failure or even necrosis. In fact, patients in intensive care units (ICUs) with elevation of troponin in the absence of coronary artery disease have a worse prognosis.10,11
Determinants of Cardiac Output
Low cardiac output from any cause impairs oxygen delivery and respiration at the cellular level. A more detailed diagnostic approach that defines the variable of cardiac output and allows for a more targeted approach to therapy is often required. Three variables determine cardiac output and distribution of blood to the periphery: stroke volume (ventricular function), heart rate (rhythm), and vascular resistance (systemic).
Arterial Pressure = cardiac output × total (systemic) vascular resistance
Cardiac output = stroke volume × heart rate
Determinants of cardiac function and other variables influencing cardiac output are complex; but one major determinant relevant to the initial assessment is the volume loading conditions of the heart. At normal volume loading conditions and in the absence of pathologic conditions, cardiac output is optimal at rest. When cardiac output falls, compensatory mechanisms are activated to maintain cardiac output. Historically, it was taught that these mechanisms in cardiogenic shock included an increase in heart and peripheral systemic vascular resistance. New data however suggest that cardiogenic shock may be heterogeneous and in some cases resemble a systemic inflammatory state (see below).
If the diagnosis or etiology of shock is unclear, a pulmonary arterial catheter can measure filling pressures (central venous and pulmonary capillary wedge pressures) and cardiac output and calculate systemic vascular resistance. With three variables, there are 27 possible hemodynamic combinations, so interpretation can be complex. But three general hemodynamic subdivisions allow better diagnostic accuracy (Table 7-1). It should be remembered that insertion of a pulmonary artery (PA) catheter is a diagnostic and not a therapeutic procedure, with associated complications. In most patients, clinical assessment of filling pressure (central venous pressure, rales) and clinical circumstances are diagnostic and sufficient.
An individualized approach to the use of a PA catheter is recommended by the American College of Cardiology (ACC)/American Heart Association (AHA) guidelines.12 There has been a decline in PA catheter use and controversy concerning an association with poor outcome,13 but no association has been shown in patients with MI and cardiogenic shock.14
Table 7-1 • Hemodynamic Parameters in the Three Major Categories of Shocka | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
Cardiogenic Shock Complicating Acute Myocardial Infarction
Cardiogenic shock is described as inadequate organ perfusion resulting from depressed cardiac function in the presence of normal intravascular volume. Infarction of 40% or more of the LV myocardium in acute STEMI usually results in cardiogenic shock and death. Although the mortality rate of cardiogenic shock has decreased in selected recent trials, death rates have averaged 50% to 70%.4 The overall incidence had not declined appreciably15,16 until recently, with increasing use of revascularization, especially primary PCI, in both the United States and Europe.17,18,19
Risk factors for the development of cardiogenic shock with MI include advancing age, anterior MI, prior MI or angina, multivessel coronary artery disease, new bundle-branch block with MI, or prior heart failure.20 Mortality varies according to demographic, clinical, and hemodynamic parameters. These include age, clinical signs of poor peripheral perfusion, anoxic/hypoxic brain damage, and left ventricular ejection fraction. Of these, hemodynamic factors are associated with short- but not long-term prognosis.
Cardiogenic Shock in NSTEMI Acute Coronary Syndrome
It should also be appreciated that shock can occur in patients with non–ST-elevation MI (NSTEMI), and even a small or modest infarct can cause hemodynamic instability if prior MI or LV dysfunction is present at the time of recurrent ACS. The SHOCK trial found that approximately 17% of all cardiogenic shock complicating MI was associated with NSTEMI. Patients with NSTEMI are significantly older and have more prior MI, heart failure, and three-vessel disease. They also have more comorbidities—including renal dysfunction, bypass surgery, and peripheral vascular disease—than patients with STEMI.5,21 The Global Use of Strategies to Open Occluded Coronary Arteries (GUSTO)-II and PURSUIT trials found that cardiogenic shock occurs in up to 5% of patients with NSTEMI and that mortality rates are >60%.5,22
Pathophysiology and Hemodynamics of Cardiogenic Shock
MI may result in hemodynamic instability and congestive heart failure (CHF). As described above, cardiogenic shock classically has been defined as a “pump” problem due to “massive” heart attack. In this context, cardiac output and ventricular ejection fraction fall due to severe left ventricular dysfunction. Heart rate increases to compensate for the fall in stroke volume in a reflex compensatory effort to maintain cardiac output. If the patient survives initially, ventricular dilation and remodeling occur over days to months, resulting in an increase in end-diastolic volume (increased ventricular preload), which may help to maintain stroke volume despite the fall in ejection fraction.
In cardiogenic shock, the degree of myocardial dysfunction is often severe. But in many case this severe impairment of contractility does not lead to shock, and LV ejection fraction (LVEF) may only be moderately depressed.23 In the SHOCK trial, mean ejection fraction was ∼30%.24 In addition, LVEF is similar in the acute phase and 2 weeks later, when shock has resolved in survivors. Approximately half of patients with cardiogenic shock have small or normal LV size representing failure of an adaptive mechanism of acute dilation to maintain stroke volume early in MI.25 Paradoxically, reduced ejection fraction and ventricular dilation are prognostic indicators of increased survival in septic shock syndrome.26
Iatrogenic Cardiac Shock
The majority of patients with cardiogenic shock following MI develop shock after the first 24 hours. In some patients at high risk for cardiogenic shock, medications contribute to the development of shock, including beta-blocking agents, angiotensin converting enzyme inhibitors (ACEIs), and other vasodilators such as nitrates and morphine27,28,29,30 (Fig. 7-1).
In acute pulmonary edema, intravascular volume is redistributed to the extravascular pulmonary interstitial space in the lungs. A tachycardia may be largely compensatory for depressed myocardial function and tenuous intravascular volume. Beta-blocking agents may decrease heart rate diminishing stroke volume and vasodilators or diuretics may further redistribute blood volume or cause a poorly tolerated diuresis, precipitating a low output state. Low-dose diuretics should be initially used and nitrates added cautiously. Noninvasive positive-pressure ventilation may be useful if tolerated. IV ACEIs are contraindicated early in MI. Oral ACEIs should be
deferred until patients are hemodynamically stable. In ISIS-4, the only significant side effect of captopril was hypotension.28 In this trial, patients who developed hypotension were at increased risk for the development of cardiogenic shock and tended to have lower blood pressures and higher heart rates.
deferred until patients are hemodynamically stable. In ISIS-4, the only significant side effect of captopril was hypotension.28 In this trial, patients who developed hypotension were at increased risk for the development of cardiogenic shock and tended to have lower blood pressures and higher heart rates.
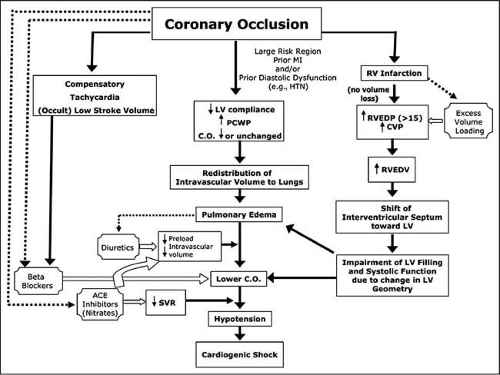 Figure 7-1 • Pathophysiology of iatrogenic shock. Acute pulmonary edema is a state of redistribution of intravascular volume into extracellular space in the lungs (center pathway). When hemodynamic stability is tenuous, the additional decrease in plasma volume caused by diuretics in patients without prior heart failure may induce shock. Tachycardia is often compensatory for lower stroke volume but is not appreciated as such (left-sided pathway). Treatment with betablockade lowers heart rate and stroke volume, leading to shock. Decompensation may also occur when patients who are reliant on compensatory vasoconstriction are aggressively treated with angiotensin-converting enzyme inhibitors, particularly intravenously and very early before they have hemodynamically stabilized. Nitrates would be expected to have a similar effect but did not in the only systematic study, which used oral low-dose treatment. Volume expansion may be deleterious when used to excess or when RV filling pressure is already elevated, because the RV may become volume overloaded with shift of the septum, causing impairment in LV filling and contraction (right-sided pathway). CVP, central venous pressure; HTN, hypertension; PCWP, pulmonary capillary wedge pressure; RVEDP, RV end-diastolic pressure; RVEDV, RV end-diastolic volume. (From Reynolds HR, Hochman JS. Circulation 2008;117[5]:686–697, with permission.) |
It is important to note that MI may lead to pulmonary edema without the development of shock. An initial effect of ischemia is a decrease in LV compliance, and pulmonary edema initially may mimic diastolic heart failure.
Beta-Blockade and Heart Failure—Updated Guidelines
As noted, tachycardia may help maintain cardiac output despite the fall in ejection fraction and stroke volume. But all compensatory changes are likely to increase myocardial oxygen consumption. They can worsen ischemia in viable or distant myocardium and extend the infarct area. In patients without acute heart failure, a reduction in heart rate with beta-blockade improves outcome mainly by decreasing episodes of fatal ventricular fibrillation. Blockade of excess sympathetic and neurohumoral stimulation reduces myocardial oxygen consumption. But in compensatory tachycardia, beta-blockade can be life-threatening, as in cardiogenic shock or severe heart failure, when the stroke volume is critically dependent on the tachycardia (Fig. 7-2) (see Chapter 5).
Hemodynamic Parameters of Cardiogenic Shock
When LV end-diastolic pressure increases substantially (>25–30 mm Hg), interstitial and then pulmonary edema develops. If RV end-diastolic pressure increases, peripheral
edema will be observed. A fall in cardiac output also triggers an adrenergic response, producing tachycardia and peripheral vasoregulatory changes that try to redistribute blood flow. Constriction of arteries to the skin, kidneys, and gut redistributes blood flow away from these tissues to maintain blood flow to the brain and heart. But this systemic vasoconstriction may create increased LV afterload, impeding LV ejection. As cardiac output continues to fall, hypotension and lactic acidosis develop. This combination of pulmonary edema with signs of inadequate systemic perfusion is the hallmark of cardiogenic shock.
edema will be observed. A fall in cardiac output also triggers an adrenergic response, producing tachycardia and peripheral vasoregulatory changes that try to redistribute blood flow. Constriction of arteries to the skin, kidneys, and gut redistributes blood flow away from these tissues to maintain blood flow to the brain and heart. But this systemic vasoconstriction may create increased LV afterload, impeding LV ejection. As cardiac output continues to fall, hypotension and lactic acidosis develop. This combination of pulmonary edema with signs of inadequate systemic perfusion is the hallmark of cardiogenic shock.
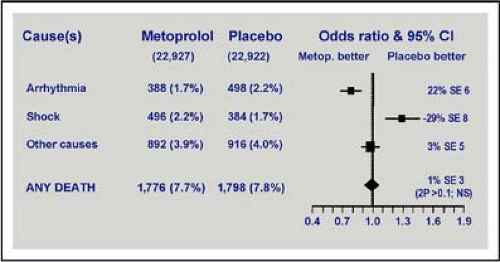 Figure 7-2 • Effects of metoprolol on death before discharge from hospital. A beneficial effect on reduction in deaths due to arrhythmia was negated by an offsetting increase in patients dying from cardiogenic shock. These deaths occurred primarily during the first day of admission. (Modified from Chen ZM, Pan HC, Chen YP, et al. Lancet 2005;366[9497]:1622–1632, with permission.) |
The patient with LV dysfunction classically has been described as one with a cardiac index (cardiac output corrected for body surface area) ≤2.2 L/min/m2, PCWP >18 mm Hg, and SBP <90 mm Hg. When the cardiac index falls to 2.2 L/min/m2 and SBP falls to 90 mm Hg, frank signs of poor peripheral perfusion are usually present.
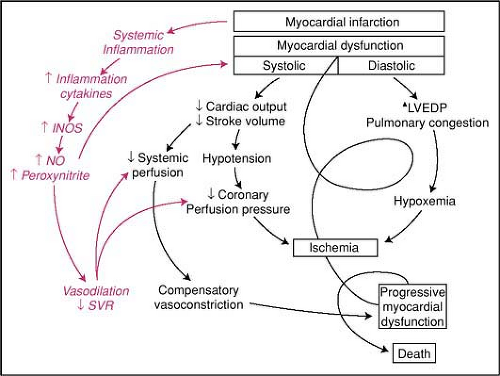 Figure 7-3 • Classic shock paradigm, as illustrated by S. Hollenberg, is shown in black. The influence of the inflammatory response syndrome initiated by a large MI is illustrated in red. LVEDP indicates left ventricular end-diastolic pressure. (From Hochman JS. Cardiogenic shock complicating acute myocardial infarction: expanding the paradigm. Circulation 2003;107:2998–3002, with permission.) |
Classic Hemodynamic Definition of Cardiogenic Shock
Cardiogenic shock is defined31 as SBP ≤90 mm Hg for ≥1 hour that is
Not responsive to fluid administration alone
Secondary to cardiac dysfunction
Associated with signs of hypoperfusion or a cardiac index ≤2.2 L/min per m2 and a pulmonary capillary wedge pressure >18 mm Hg
Changing the Paradigm of Cardiogenic Shock
Related posts:
 Emergency Department Management of Acute Coronary Syndromes
Emergency Department Management of Acute Coronary Syndromes
 Heart Failure and Acute Pulmonary Edema in the Emergency Department
Heart Failure and Acute Pulmonary Edema in the Emergency Department
 Restoring Life: The Story of Human Defibrillation and Modern CPR
Restoring Life: The Story of Human Defibrillation and Modern CPR
 Oxygen Administration and Supraglottic Airways
Oxygen Administration and Supraglottic Airways
 Life-Threatening Arrhythmias: Evaluation, Identification, and Assessment
Life-Threatening Arrhythmias: Evaluation, Identification, and Assessment

Full access? Get Clinical Tree


