Chapter 8
Cardiogenic Shock and Other Pump Failure States
Cardiogenic shock occurs when impairment of cardiac pump function results in inadequate tissue perfusion. This chapter focuses on the pathophysiology, clinical diagnosis, and approach to the patient with cardiogenic shock; Chapter 9 discusses shock resulting from low preload, and Chapter 10 discusses shock resulting from the maldistribution of blood flow. Pericardial tamponade and major pulmonary embolus, the two primary causes of obstructive shock, are presented in Chapters 54 and 77, respectively. Chapter 52 addresses acute heart failure syndromes that overlap with cardiogenic shock with regard to specific etiologies.
Pathophysiology
Determinants of Tissue Perfusion
The principal determinants of tissue perfusion are cardiac output and arterial blood pressure. Cardiac output is defined by the relationship in Equation 1 (Box 8.1). Factors that affect ventricular stroke volume include preload, intrinsic myocardial contractility, and afterload (Figures 8.1 to 8.4). Arterial blood pressure represents the driving force for tissue perfusion and can be defined by Equations 2 and 3 (see Box 8.1). Systemic vascular resistance is principally determined by the arterioles. Shock can be caused by a variety of pathophysiologic processes that alter any of these factors, thereby reducing oxygen delivery to the tissues, and these can be organized by hemodynamic alteration as shown in Table 8.1.
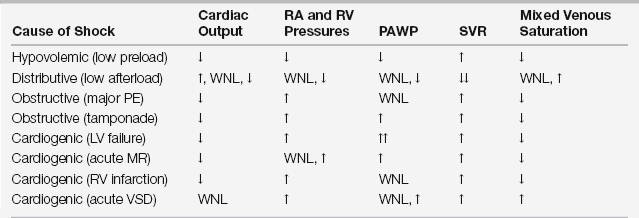
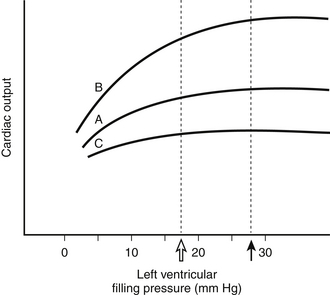
Figure 8.1 “Starling curves” of ventricular function representing the relationship between cardiac output (as the dependent variable) and left ventricular filling (end-diastolic) pressure (LVEDP) (as the independent variable) for myocardial states of normal (A), enhanced (B), and decreased (C) myocardial contractility. Other important independent variables that determine cardiac output, such as afterload (see Figure 8.3), are held constant. In the intensive care unit, LVEDP is normally approximated by pulmonary artery wedge pressure (PAWP). The dashed vertical line at ~18 mm Hg (open arrow) indicates the PAWP at which fluid begins to accumulate in the interstitial space of the lung. The dotted vertical line at ~28 mm Hg (closed arrow) indicates the PAWP at which acute alveolar edema develops. Note that all curves lack “descending limbs” at high filling pressures (i.e., decreasing cardiac outputs at high LVEDP). Descending limbs of Starling curves are considered to be experimental artifacts and may have been due to development of mitral regurgitation at high distending pressures. (See Elzinga G: Starling’s “Law of the heart”: Rise and fall of the descending limb. News Physiol Sci 7:134-137, 1992, for more details about the descending limb.)
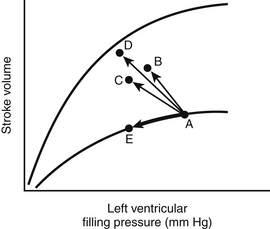
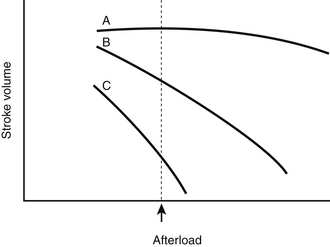
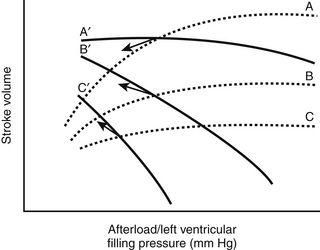
Figure 8.4 Three Starling curves (dotted lines) relating stroke volume and left ventricular filling pressure with normal (A), moderately decreased (B), and severely decreased (C) myocardial function (Figure 8.1) are superimposed on three curves (solid lines) relating stroke volume and afterload for the same three states of myocardial function (normal, A′; moderately decreased, B′; and severely decreased, C′) (Figure 8.3). Because most vasodilator agents (e.g., nitroprusside) reduce both preload and afterload, their effects on stroke volume depend on the state of myocardial function. For example, when myocardial function is normal, such a vasodilator lowers stroke volume because of the predominant effect caused by lowering preload (as shown by the arrow originating at the intersection of curves A and A′). In contrast, when myocardial function is depressed, such an agent results in improved stroke volume despite a decrease in preload (arrows from the intersections of curves B and B′ and curves C and C′) (similar to point C in Figure 8.2). (Modified from Cohn JN, Franciosa JA: Vasodilator therapy of cardiac failure. N Engl J Med 297:27-31, 1977.)
Stages of Shock
The shock syndrome is characterized by a series of physiologic stages beginning with an initial inciting event that causes acute circulatory compromise. Shock may subsequently progress through three stages, culminating in irreversible end-organ damage and death.
Preshock
Preshock is also known as compensated shock. During this stage, the body’s homeostatic mechanisms rapidly compensate for diminished perfusion. Reflex sympathetic activation leads to tachycardia and peripheral vasoconstriction, thereby temporarily maintaining blood pressure and cardiac output.
Frank Shock
During this stage, the regulatory mechanisms become overwhelmed, and signs and symptoms of organ dysfunction begin to appear, including tachycardia, tachypnea, metabolic acidosis, and oliguria. The emergence of these signs typically corresponds to one or more of the following: (1) a 25% reduction in effective blood volume in hypovolemic shock, (2) a decrease in the cardiac index to less than 2.5 L/min/M2, or (3) activation of the many mediators of the sepsis syndrome (Chapter 10).
Irreversible Shock
During this stage, progressive end-organ dysfunction leads to irreversible organ damage and eventual death: (1) urine output may decline and renal failure may ensue; (2) mental status may become altered, with agitation, obtundation, and eventually coma; (3) respiratory muscle fatigue may be precipitated by decreased perfusion of the diaphragms, leading to hypercapnic respiratory failure; and (4) multiple organ system failure may ensue.
Differential Diagnosis
To initiate appropriate therapy, cardiogenic shock must be differentiated from other categories of shock, such as hypovolemic shock, distributive (low afterload) shock, or obstructive shock. The clinical history, physical examination, and laboratory finding should provide important clues to the shock state’s origin. The use of a balloon-tipped, flow-directed pulmonary artery (Swan-Ganz) catheter can facilitate the initial categorization of the shock state as well as help to identify the individual causes of cardiogenic shock (see Table 8.1 and Box 8.2). Echocardiography with Doppler can provide similar, although more limited assessment of these parameters as well.
Shock caused by vasodilation or low afterload is termed distributive shock (e.g., shock caused by sepsis or anaphylaxis). Septic shock occurs as a result of endogenous and exogenous biologically active factors, which produce vasodilation and impair oxygen delivery to the tissues. Early septic shock may be associated with increased cardiac output secondary to decreased afterload and increased heart rate, but late septic shock may be associated with profound reduction in cardiac output from myocardial depression resulting from a number of factors. Late septic shock needs to be distinguished from primary cardiogenic shock because the therapy for these conditions differs significantly.




