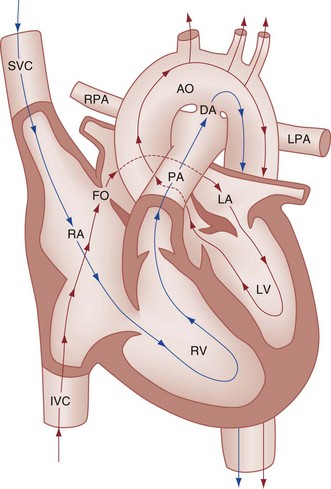
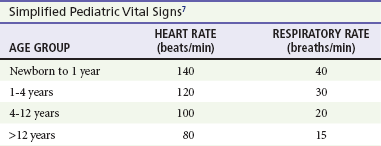
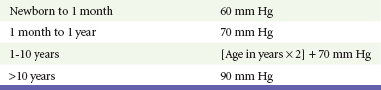
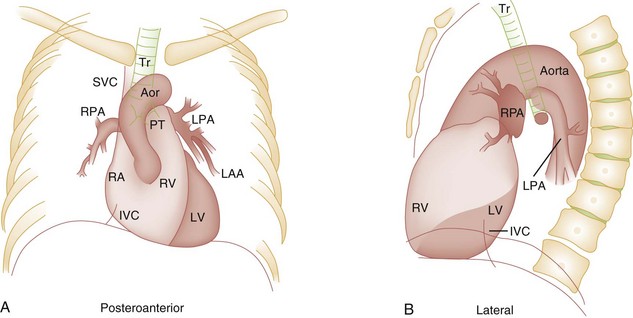
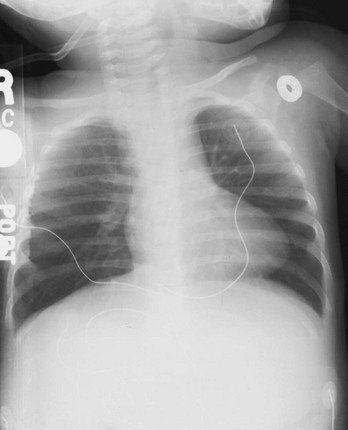
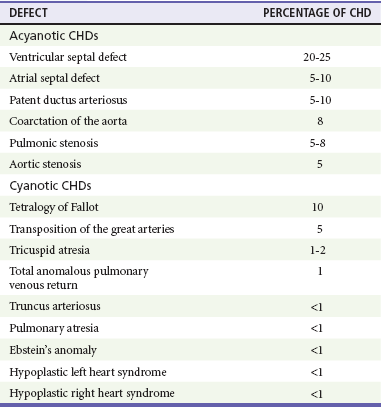
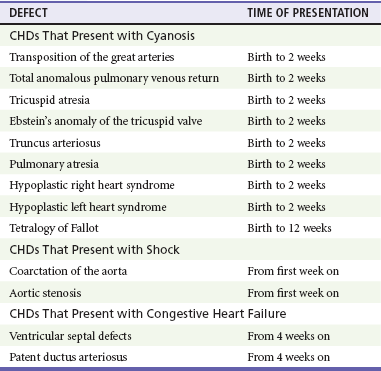
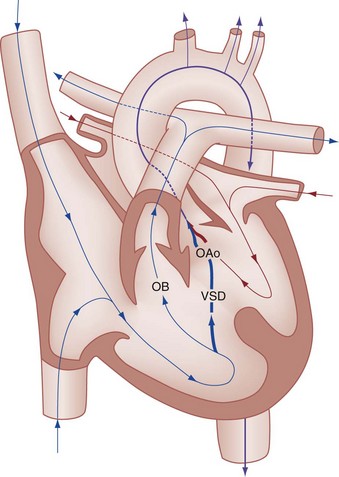
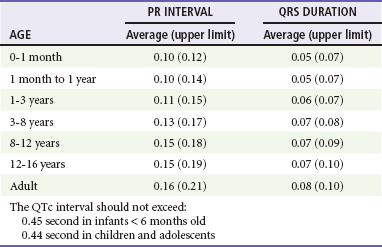
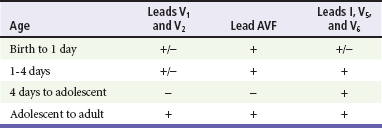
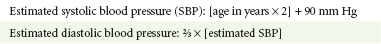Chapter 171 The second scenario represents more of a challenge to the emergency physician: the child with an undiagnosed congenital or acquired cardiac disorder who presents to the emergency department with nonspecific or concerning signs and symptoms (Box 171-1). This chapter focuses on some of the more common and life-threatening cardiac disorders in infants and children who present to the emergency department, with an emphasis on rapid evaluation, stabilization, and management of these disorders. Fetal and Neonatal Circulation Some of the key features of the fetal circulation that differ from the circulation of a child are the ductus venosus, the ductus arteriosus, and a patent foramen ovale. During fetal development, blood is oxygenated by the placenta, flows to the fetus through the umbilical vein, bypasses the fetal liver through the ductus venosus, and returns to the fetal heart through the inferior vena cava. Blood returning from the inferior vena cava then enters the right atrium and is preferentially shunted across to the left atrium through the patent foramen ovale (Fig. 171-1). The blood in the left atrium is then pumped from the left ventricle to the aorta. The oxygenated blood ejected through the ascending aorta is preferentially directed to the fetal coronary and cerebral circulations. Deoxygenated blood that returns to the right atrium through the superior vena cava crosses the tricuspid valve and is pumped into the fetal pulmonary artery through the right ventricle. Because the fetal pulmonary vascular resistance (PVR) is higher than the fetal systemic vascular resistance (SVR), this deoxygenated blood bypasses the nonoxygenated fetal lungs through the patent ductus arteriosus (see Fig. 171-1). This poorly oxygenated blood enters the aorta through the patent ductus arteriosus and then mixes with the well-oxygenated blood in the descending aorta. The mixed blood in the descending aorta then returns to the placenta for oxygenation through the two umbilical arteries. The young myocardium is inefficient and unable to increase its contractility in response to demand.1 When more cardiac output is needed, infants and children respond with an increase in heart rate. Therefore, bradycardia in infants and young children is an ominous sign that connotes a severely compromised cardiac output. Children develop the adult capacity to increase their stroke volume to improve overall cardiac output by 8 to 10 years of age.2 On the basis of the first physiologic formula, as stroke volume decreases, a compensatory increase in the heart rate will be necessary to preserve a normal cardiac output. A decrease in stroke volume can be produced by a weak “pump,” decreased volume in the circulation, or both. The most common cause of decreased stroke volume in children is hypovolemia due to dehydration. Other causes of decreased stroke volume in children may be implicated (Box 171-2). Thus tachycardia is the first compensatory cardiovascular response when the stroke volume is decreased. If tachycardia alone is not enough to maintain a normal cardiac output, the next compensatory physiologic mechanism to preserve perfusion is an increase in the SVR. This increase in SVR is exhibited as an increase in the diastolic blood pressure, which in turn accounts for a narrowed pulse pressure. The clinical examination findings of the extremities of a child with an increased SVR include pallor, mottling, cool skin, delayed capillary refill time (>2 seconds), and weak or thready distal pulses. Cyanosis is a clinical sign caused by the presence of deoxygenated blood in the capillary beds, most readily observed in the mucous membranes, conjunctiva, nail beds, and skin. The presence of cyanosis usually means that there are at least 4 to 5 g/dL of deoxyhemoglobin in the blood, which correlates with an oxygen saturation of about 80 to 85%.3 Central cyanosis results from a decrease in pulmonary ventilation and oxygenation, a decrease in pulmonary perfusion, the shunting of deoxygenated blood directly into the systemic circulation, or the presence of abnormal hemoglobin. Cyanosis in the neonate can be due to a variety of cardiac, pulmonary, or hematologic causes. Cardiac causes of cyanosis include congenital lesions with right-to-left shunts and cardiac lesions with decreased or increased pulmonary blood flow. Common pulmonary causes of cyanosis include bronchiolitis, pneumonia, and pulmonary edema. Methemoglobinemia can be one of the hematologic causes of cyanosis. The region of the body that is cyanotic can provide important clinical clues to the cause of the cyanosis. Central cyanosis involves the lips, tongue, and mucous membranes, whereas peripheral cyanosis (acrocyanosis) involves the hands and feet. Acrocyanosis is a common phenomenon in neonates caused by cold stress and peripheral vasoconstriction. Central cyanosis reflects a pathologic origin and is an ominous sign. Infants with cyanosis secondary to a congenital heart defect may not exhibit as much respiratory distress compared with the infant with cyanosis due to a pulmonary cause. Thus a cardiac cause of central cyanosis may be more likely than a purely pulmonary cause in the child who appears “comfortably blue.” Another important clinical clue to the cause of central cyanosis is that cyanosis of cardiac origin usually worsens with crying, whereas cyanosis due to a pulmonary cause may improve when the infant cries.4 Cyanotic congenital heart defects with right-to-left shunting will demonstrate a minimal improvement with supplemental oxygen, whereas cyanosis of a purely pulmonary origin typically exhibits a significant improvement with supplemental oxygen (Table 171-1). Table 171-1 Clinical Clues to Help Distinguish between Cardiac and Pulmonary Causes of Central Cyanosis The key elements that should be elicited in the history of a child with a known underlying cardiac disorder are listed in Box 171-3. Early consultation with the child’s cardiologist or cardiac surgeon is extremely useful. Infants with an underlying congenital heart disorder may exhibit diaphoresis during feeds and poor weight gain secondary to congestive heart failure (CHF). The cause of the infant’s hypoxia—cardiac or pulmonary—may be ascertained by the age at onset and the events surrounding a change in color. For example, an infant who sweats during feeding may exhibit a splanchnic steal from anomalous coronary arteries, causing transient ischemia, pain, color change, and diaphoresis that resolve after eating.1 A child with an undiagnosed congenital heart defect may take longer to feed, frequently pausing to catch his or her breath, with subsequent poor weight gain and gradually increased work of breathing due to having CHF and pulmonary edema. Respiratory tract infections are common during childhood and may cause an acute deterioration in a child with an underlying cardiac disorder. In turn, children with congenital heart disease (CHD) with large left-to-right shunts and increased pulmonary blood flow tend to have a higher incidence of lower respiratory tract infections. Acute respiratory distress in these patients may be from a combination of pulmonary and cardiac factors (e.g., CHF). The majority of pediatric chest pain cases are noncardiac in origin and benign in nature. Common causes include musculoskeletal or chest wall pain, costochondritis, asthma exacerbations, pneumonia, pleurisy, gastritis, and gastroesophageal reflux. An often underdiagnosed cause of acute chest pain in the child or adolescent is referred to as the precordial catch syndrome (also known as Texidor’s twinge). The pain is sharp, nonradiating, and located in the left periapical area of the chest wall; it occurs suddenly and is often exacerbated during inspiration but is not associated with dyspnea.5 Patients may report that the pain took their breath away or that they were afraid to move; it typically resolves within a few minutes and is not associated with dysrhythmias or other sequelae. The pathophysiologic mechanism of precordial catch syndrome is unknown; the pain may originate from the parietal pleura or costal cartilage.5 If an ECG and a chest radiograph are performed, findings are normal in these patients. The pain is fleeting but may recur at any time and at any age; giving the patient’s symptoms a name emphasizes certainty in and familiarity with the diagnosis and reassures family members concerned about a cardiac etiology of their child’s chest pain.6 A mild resting tachypnea or tachycardia may be the only clinical clue to an underlying cardiovascular disorder. Age-related variables in heart rates, respiratory rates, and blood pressures can be a source of frustration and confusion to those clinicians who do not manage pediatric patients on a routine basis. Although there are numerous tables of pediatric vital signs with variations based on sleep or awake states, one can easily recall a rough estimate of the normal pediatric heart rates and respiratory rates by a simplified table of pediatric vital signs (Table 171-2).7 The methods to calculate the normal expected blood pressures and hypotensive blood pressures are also listed in Table 171-2.8 An accurate blood pressure reading is accomplished by use of a cuff that covers two thirds of the upper arm or thigh. A cuff that is too narrow will overestimate the patient’s true blood pressure, and a cuff that is too large will underestimate the true blood pressure. Any child with a suspected cardiac disorder should have blood pressures measured in both arms. If the blood pressure in the left arm is significantly lower than the blood pressure in the right arm, a coarctation of the aorta proximal to the origin of the left subclavian artery should be suspected. Blood pressures should be measured in the thighs in any child with a suspected aortic coarctation or documented hypertensive blood pressures in the upper extremities. The mere presence of femoral pulses does not rule out clinically the possibility of a coarctation of the aorta. Even with an appropriately sized cuff, the blood pressures in the thighs can be 10 to 20 mm Hg higher than the blood pressures in the upper extremities because of the lack of well-designed blood pressure cuffs for the legs. Therefore, if the measured blood pressure in the lower extremities is lower than the blood pressure in the upper extremities, coarctation of the aorta should be suspected. Pulse oximetry readings that are lower in the legs than in the upper extremities are also suggestive of either a coarctation of the aorta or a right-to-left-shunt across a patent ductus arteriosus.9 Cardiac murmurs are produced by turbulent blood flow through the heart. The presence of a cardiac murmur may not be associated with an underlying cardiac defect, however. The location, intensity, quality, timing, and radiation of the murmur determine whether the murmur is suggestive of an underlying cardiac pathologic condition. Although systolic murmurs can be present without any underlying anatomic abnormalities, diastolic murmurs are always considered pathologic in nature. Some of the other criteria that would suggest an underlying anatomic cardiac abnormality are listed in Box 171-4. Murmurs may be difficult to appreciate in the noisy emergency department setting and given the degree of tachycardia that is often present even in normal infants. However, the location of the murmur may be a valuable clinical tool in determining the underlying anatomic origin of the murmur (Box 171-5). The hyperoxia test is an important bedside diagnostic tool to help differentiate between cardiac and pulmonary causes of central cyanosis. This test consists of assessment of the rise in arterial oxygenation with the administration of 100% oxygen. An arterial blood gas is measured on room air (if tolerated) and repeated after several minutes on high-flow oxygen (100% oxygen); the two blood gas analyses are then compared. When the child is breathing high-flow oxygen, an arterial oxygen partial pressure (PaO2) of more than 250 mm Hg virtually excludes hypoxia due to CHD—a “passed” hyperoxia test.10 An arterial oxygen reading of less than 100 mm Hg (in a child without obvious pulmonary disease) is consistent with a right-to-left shunt and is highly predictive of CHD—a “failed” hyperoxia test.10 Values between 100 and 250 mm Hg may indicate lesions with intracardiac mixing. Pulse oximetry is not an appropriate substitute for an arterial blood gas analysis; it is not sensitive enough to determine “pass or fail” of the test because a child breathing high-flow oxygen and registering 100% on pulse oximetry may actually have a PaO2 anywhere between 80 and 680 mm Hg.1 Prolonged administration of 100% oxygen may cause some theoretic problems, such as closure of the ductus arteriosus in those infants with critical left-sided heart obstructions or pulmonary vasodilation (which would potentially worsen pulmonary vascular congestion).3 However, oxygen should not be withheld initially in critically ill infants; continuous reassessment is crucial in the evaluation and management of children with suspected CHD.1 Three features of the chest radiograph (Fig. 171-2) that deserve special attention are the cardiac size (cardiothoracic ratio), the cardiac shape (silhouette), and the degree of pulmonary vascular markings. The easiest method to determine the heart size in children is to determine the cardiothoracic ratio, which is obtained by comparing the largest transverse diameter of the cardiac shadow on the posteroanterior view of the chest radiograph with the widest internal diameter (measured from the inside rib margin to the widest point above the costophrenic angles) of the chest. The films should be obtained during maximal inspiration whenever possible. The normal cardiothoracic ratio in children is approximately 50%. The cardiothoracic ratio is not very accurate in newborns and small infants, in whom a good inspiratory view is rarely obtained.11 A cardiac silhouette that is larger than normal may be due to a shunt lesion, cardiomegaly, or pericardial effusion.11 An enlarged heart shadow on a chest radiograph more reliably reflects a problem with volume overload rather than pressure overload. Problems with pressure overload are better represented on the ECG. The cardiac size can be falsely increased in infants by the presence of the thymus, which can be seen in the mediastinum on the chest radiograph from birth until about 5 years of age. The thymic borders are typically wavy in appearance and sometimes can be seen as the classic “sail sign” along the superior right border of the heart (Fig. 171-3). The thymic shadow may not be visible radiographically in infants during times of physiologic stress but should reappear when the infant recovers. Figure 171-3 Thymic shadow demonstrating the “sail sign” along the right cardiac border (dotted line). The three classic cardiac silhouettes seen in patients with congenital heart defects are the boot-shaped heart of tetralogy of Fallot (Fig. 171-4), the egg-on-a-string silhouette of transposition of the great vessels, and the snowman-shaped or figure-of-eight heart of total anomalous pulmonary venous return. In a normal left-sided aortic arch, the aorta descends to the left of the midline and displaces the tracheal air shadow slightly toward the right of midline above the level of the carina. In contrast to this, the tracheal air shadow may be midline or deviated toward the left in the presence of a right-sided aortic arch.3 It is important to note this finding because a right-sided aortic arch is found in up to 25% of the children with tetralogy of Fallot.9 Rib notching secondary to increased collateral blood flow along the intercostal vessels can sometimes be appreciated between the fourth and eighth ribs in older children with undiagnosed coarctation of the aorta but is rarely visualized in children with coarctation of the aorta who are younger than 5 years.11 The electrocardiographic findings in infants and children can sometimes be problematic because various components of the ECGs change according to the child’s age (Table 171-3).8 At birth, the muscle mass of the right ventricle is greater than that of the left ventricle; this is demonstrated by right axis deviation on the neonatal ECG. By the end of the first month of life, the left ventricle assumes dominance. By 6 months of age, the left ventricular to right ventricular mass ratio is 2:1, which then reaches the adult ratio of 2.5:1 by adolescence. The durations of the PR interval, QRS complex, and QT intervals increase with age. Table 171-3 Normal Electrocardiographic Values (PR, QRS, QTc, and QRS Axes) in Infants and Children8 As in adults, cardiac troponin T (cTnT) and cardiac troponin I (cTnI) are highly sensitive and specific in children for myocardial damage.11 Reference values are slightly higher for neonates younger than 3 months; normal and indeterminate values will depend on the bioassay used.11 The indications for troponin testing in children include suspected cardiac ischemia (of any etiology), myocarditis, and myocardial dysfunction in sepsis syndrome.11,12 Several studies have evaluated the use of plasma B-type natriuretic peptide (BNP) levels in the assessment and management of CHF in adults.13 Studies of BNP levels in children have demonstrated a similar correlation of elevated levels in children with CHF, and these also correlated with the clinical symptoms of heart failure and the ejection fraction as measured by echocardiography.14 The clinician is urged to refer to the particular range of age-specific values from the laboratory kit used at his or her institution. The incidence of CHD in the United States has remained fairly constant at approximately 1%, or 8 to 10 cases per 1000 live births. This equates to approximately 32,000 infants born each year with some form of CHD15 (Table 171-4). Although a large percentage of CHD is now detected with prenatal ultrasonograms, one study also recommended routine pulse oximeter readings in all newborns before discharge from the nursery as an additional inexpensive screening tool for CHD.16 The age, severity of symptoms, and time of presentation of a child with CHD vary by the specific defect, complexity and severity of the defect, and timing of the normal physiologic changes that occur as the fetal circulation transitions to that of a neonate (Table 171-5). The more severe or complex CHD lesions may not be clinically apparent immediately after birth. However, as the ductus arteriosus begins to close in the first several weeks of life, cardiac defects with obstructive lesions of the pulmonary or systemic circulations will be unmasked, and these infants will present clinically with acute cyanosis, shock, or both. Even the harsh systolic murmur of a large, isolated ventricular septal defect may not be heard until about the fourth to sixth week of life when the left-to-right shunt across the ventricular septal defect increases because of the decrease in the PVR. In general, the more severe the anatomic defect is (i.e., lack of pulmonary blood flow or lack of systemic blood flow), the earlier in life these conditions will be manifested with cyanosis and shock. The emergency physician must rely on several key elements of the clinical examination in addition to findings on the chest radiograph and ECG to narrow the diagnostic possibilities. For example, with use of the data in Box 171-6, the presence of cyanosis, a grade 3/6 systolic ejection murmur best heard at the midleft sternal border, a boot-shaped heart, and a decreased pulmonary blood flow on the chest radiograph with evidence of right ventricular hypertrophy on the ECG suggest tetralogy of Fallot. Only a brief discussion of some of the more common CHDs is presented in this chapter. One unique pharmacologic intervention that can be lifesaving in infants involves the use of prostaglandin E1 (PGE1) to maintain the patency of the ductus arteriosus. CHD that is manifested within the first 2 to 3 weeks of life with a sudden onset of cyanosis or cardiovascular collapse is typically due to duct-dependent cardiac lesions (Box 171-7).4 Closure of the ductus arteriosus in patients with these specific cardiac lesions causes life-threatening situations by either an interruption of blood flow to the lungs, producing cyanosis (i.e., tricuspid atresia), or a disruption of blood flow to the systemic circulation, producing shock (i.e., hypoplastic left heart syndrome).4 Box 171-8 details one suggested method to prepare this potentially lifesaving PGE1 infusion. The PGE1 infusion is typically started at 0.05 to 0.1 µg/kg/min (the method of preparation described in Box 171-8). A known adverse reaction to a PGE1 infusion is apnea; one should perform endotracheal intubation on these infants before the initiation of the PGE1 infusion. Not only will intubation provide a secure airway, but controlled ventilation will also help decrease the infant’s work of breathing, shunting much needed cardiac output and metabolic demands from the overtaxed respiratory apparatus. Other adverse reactions to a PGE1 infusion include fever, seizures, bradycardia, hypotension, flushing, and decreased platelet aggregation. Acyanotic CHD can be further subdivided (Fig. 171-5) into obstructive lesions (i.e., pulmonic stenosis, aortic stenosis, and coarctation of the aorta) and lesions characterized by left-to-right shunts with an associated increase in pulmonary blood flow (i.e., ventricular septal defects, atrial septal defects, patent ductus arteriosus, and endocardial cushion defects). These acyanotic lesions usually are manifested within the first 6 months of life with symptoms of CHF; however, atrial septal defects can remain asymptomatic until adulthood. Perspective.: Ventricular septal defects are the most common congenital cardiac defects and account for 20 to 25% of all cases of CHD. Spontaneous closure occurs in 30 to 40% of all ventricular septal defects overall and in 50 to 70% of smaller ventricular septal defects.17 Clinical Features.: The degree of symptoms is dependent on the size of the ventricular septal defect and the degree of PVR. Most ventricular septal defects are clinically asymptomatic (minimal or no left-to-right shunting) immediately after birth because of the high PVR. When the PVR decreases to the normal levels at 6 to 8 weeks after birth, left-to-right shunting can then occur, and the typical systolic murmur of a ventricular septal defect will then be appreciated. Small ventricular septal defects may remain completely asymptomatic throughout childhood. Approximately 10% of the infants with large ventricular septal defects will eventually have signs and symptoms of CHF (e.g., poor feeding and poor growth) by 2 to 3 months of age because of the increased pulmonary blood flow. Older children with ventricular septal defects may exhibit signs of decreased exercise tolerance and recurrent pulmonary infections. If moderate to large ventricular septal defects are not surgically corrected, irreversible changes in the pulmonary vasculature may begin to occur as early as 6 to 12 months of age, which will result in an elevation of the PVR and pulmonary hypertension. This in turn can lead to a reversal of the shunt direction across the ventricular septal defect to now become a right-to-left shunt, Eisenmenger’s syndrome, with resultant cyanosis. Diagnostic Strategies.: The chest radiograph in children with small ventricular septal defects may be entirely normal, but cardiomegaly with increased pulmonary vascular markings is usually present with untreated moderate to large ventricular septal defects. The ECG of moderate-sized ventricular septal defects typically reveals left ventricular hypertrophy, but biventricular hypertrophy may be present in ventricular septal defects with large left-to-right shunting. Management.: All ventricular septal defects, regardless of the size of the defect, are at risk for bacterial endocarditis because of the high velocity of turbulent blood flow through them. Traditional closure of ventricular septal defects required open heart surgery. Today, however, a transcatheter closure technique that avoids the inherent risks and complications of open heart surgery and cardiopulmonary bypass has supplanted traditional methods.18–20 Perspective.: Atrial septal defects account for 5 to 10% of all cases of CHD. The majority of infants and children with atrial septal defects remain clinically asymptomatic until adulthood. Spontaneous closure has been reported in up to 40% of the cases within the first 5 years of life.17 Clinical Features.: Large atrial septal defects or those associated with comorbid conditions, such as bronchopulmonary dysplasia, can be manifested with symptoms of CHF and pulmonary overcirculation (e.g., dyspnea with feedings, poor weight gain, and frequent lower respiratory tract infections).3 The majority of atrial septal defects are discovered when a suspicious murmur is detected on a routine physical examination. A widely split and fixed S2 is a characteristic finding of atrial septal defects. Diagnostic Strategies.: The chest radiograph of children with atrial septal defects will reveal varying degrees of cardiomegaly, right atrial and right ventricular enlargement, and a prominent main pulmonary artery segment and increased pulmonary vascular markings. The ECG will reveal varying degrees of right axis deviation and right ventricular hypertrophy. All patients with unrepaired atrial septal defects will have symptoms if pulmonary hypertension ensues. Patients with large atrial septal defects that are not detected and repaired are at risk for development of Eisenmenger’s syndrome. Unlike ventricular septal defects, uncomplicated atrial septal defects are not associated with high risk of bacterial endocarditis because of the lower turbulence and velocity of blood flow through the atrial septal defects. Management.: The traditional closure of atrial septal defects, like that of ventricular septal defects, required open heart surgery to place a patch over the septal defect. Newer therapies involving closures with septal occluder devices placed by the transcatheter approach have been described.21,22 Antiplatelet therapy during the 6-month period after placement of the device is typically given and is safe and effective in preventing thrombus formation on the surface of the septal occluder device. Eisenmenger’s Syndrome.: Eisenmenger’s syndrome can occur in any large left-to-right shunt defect that is not surgically corrected. When large ventricular septal defects and atrial septal defects are not surgically corrected, irreversible changes can occur in the pulmonary arterioles, leading to pulmonary vascular obstruction and pulmonary hypertension. As the degree of pulmonary hypertension increases, the PVR may then begin to exceed the SVR. This causes right-sided pressures to exceed those on the left, causing right-to-left shunting. The reversal in the direction of shunt flow produces cyanosis. Other clinical features of patients who have Eisenmenger’s syndrome include chest pain, dyspnea on exertion, and hemoptysis.23 Perspective.: Coarctation of the aorta accounts for approximately 8% of all CHD, and up to 50% of patients with coarctation of the aorta also have an associated bicuspid aortic valve.24 The area of coarctation can occur proximal to the insertion of the ductus arteriosus (preductal type) or distal to the insertion of the ductus arteriosus (postductal type). The majority of cases (89%) are of the postductal type.24 Clinical Features.: The severity of the symptoms and the age at time of presentation are dependent on the location of the coarctation, the degree of narrowing, and the presence of any other associated cardiac defects. Infants with the rarer, preductal type of coarctation of the aorta may also exhibit differential cyanosis if the ductus arteriosus remains open.25 The upper half of the body is perfused with well-oxygenated blood supplied by the left ventricle and the ascending aorta. However, the lower half of the body will appear cyanotic, as it is largely perfused by right-to-left shunting of deoxygenated blood from the patent ductus arteriosus into the descending aorta. Infants with the preductal type of coarctation of the aorta will present with signs of circulatory failure and shock when the ductus arteriosus begins to close. The astute clinician may find a “brachial-femoral delay” by palpating both pulses simultaneously.26 Most of the asymptomatic cases of the more common postductal coarctation of the aorta are diagnosed as a result of a cardiology referral for a systolic murmur or a hypertension workup, but infants with severe postductal coarctation of the aorta can also present during the first few weeks of life with signs of circulatory failure and shock. If a child is discovered to have hypertension on a routine physical examination, it is necessary to obtain blood pressure measurements in the lower extremities to assess the possibility of coarctation of the aorta. A systolic blood pressure in the right arm that is 15 to 20 mm Hg higher than that in the legs is sufficient evidence to suspect coarctation of the aorta because the systolic blood pressure in the legs is normally higher than that in the arms.25 If the systolic pressure in the right arm is higher than that in the left arm, the area of coarctation is probably preductal and located proximal to the origin of the left subclavian artery. In general, diastolic blood pressures are similar in the upper and lower extremities. Diagnostic Strategies.: The chest radiograph will most often reveal a normal-sized cardiac silhouette and normal pulmonary vascular markings, but notching along the lower borders of the posterior fourth to eighth ribs due to the pressure of the dilated collateral vessels may be exhibited in children older than 5 years. The absence of rib notching, however, does not rule out the possibility of coarctation of the aorta. The ECG typically reveals a left axis and left ventricular hypertrophy. Suspected cases of coarctation of the aorta should be imaged with transthoracic echocardiography or cardiac magnetic resonance imaging to confirm and to define the coarctation26; in stable patients, this can be done on an outpatient basis. Management.: Definitive surgical repair of coarctation of the aorta involves resection of the narrowed section of the aorta with an end-to-end anastomosis. Complications of undiagnosed cases are related to the resultant hypertension and can include heart failure, hypertensive encephalopathy, and intracranial hemorrhages. Cyanotic CHDs are a result of either decreased pulmonary blood flow to the lungs or right-to-left shunting of desaturated blood directly into the systemic circulation. They can be further subdivided into those conditions with an increased pulmonary blood flow and those lesions with decreased pulmonary blood flow (Fig. 171-6). The classic cyanotic CHD can be remembered by the five t‘s: truncus arteriosus, transposition of the great vessels, tricuspid atresia, tetralogy of Fallot, and total anomalous pulmonary venous return. Other forms of cyanotic CHD include Ebstein’s anomaly, pulmonary atresia, severe pulmonary stenosis, hypoplastic left heart syndrome, and hypoplastic right heart syndrome. Many of these cyanotic heart lesions are routinely detected either on prenatal ultrasonographic examinations or in the nursery; only tetralogy of Fallot is covered in this section. Perspective.: Tetralogy of Fallot accounts for approximately 10% of all cases of CHD and is the most common cause of cyanotic CHD beyond infancy. Tetralogy of Fallot is often associated with other cardiac defects, such as right-sided aortic arch (25% of patients), atrial septal defect (10% of patients), and anomalous origin of the left coronary artery.27 Tetralogy of Fallot arises from a single embryologic defect in which the subpulmonic conus fails to expand, resulting in the four abnormalities (Fig. 171-7): (1) right ventricular outflow tract obstruction; (2) large, unrestrictive, malaligned ventricular septal defect; (3) over-riding aorta that receives blood flow from both ventricles; and (4) right ventricular hypertrophy secondary to the high pressure load placed on the right ventricle by the right ventricular outflow tract obstruction. These anatomic defects collectively result in decreased pulmonary blood flow and varying degrees of right-to-left shunting of deoxygenated blood across the ventricular septal defect.
Cardiac Disorders
Perspective
Principles of Disease
Pathophysiology of Cardiovascular Compensatory Responses
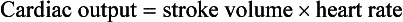
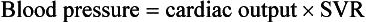
Pathophysiology of Cyanosis
Clinical Features of Cyanosis
CARDIAC ETIOLOGY
PULMONARY ETIOLOGY
Respiratory status
May be “comfortably blue”
Respiratory distress
Response to crying
Worsening cyanosis
Improved cyanosis
Response to oxygen
Minimal or no improvement
Improvement with oxygen
Clinical Features and Diagnostic Strategies: the Cardiac Evaluation
History
Chest Pain
Physical Examination
Vital Signs and Blood Pressures
Cardiac Auscultation
Hyperoxia Test
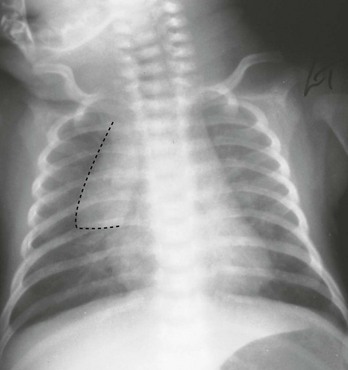
Chest Radiography
Electrocardiography

Biochemical Markers
Specific Disorders
Perspective
Clinical Features
Diagnostic Strategies
Management
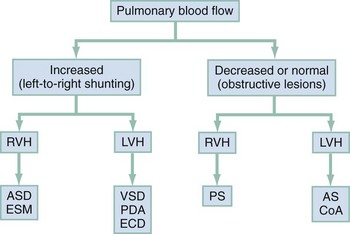
Acyanotic Congenital Heart Defect
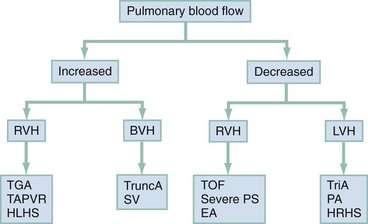
Cyanotic Congenital Heart Diseases
< div class='tao-gold-member'>

Full access? Get Clinical Tree