Cardiac Anatomy and Physiology
Paul S. Pagel
John P. Kampine
David F. Stowe
Key Points
Related Matter
Coronary Blood Flow
Laplace
Pressure–Volume Loop
Introduction
The heart is a phasic, variable speed, muscular pump that is electrically self-sustaining and provides its own blood supply. The two pair of atria and ventricles of the heart are elastic chambers that are arranged to supply equal amounts of blood to the pulmonary and systemic vasculature. Atrial and ventricular myocardium responds to stimulation rate and muscle stretch before (preload) and after (afterload) contraction begins. Coronary arterial blood vessels supply oxygen and metabolic substrates to the heart. The mechanical characteristics of the myocardium and its response to changes in autonomic nervous system activity allow the heart to adapt to rapidly changing physiologic requirements. The contractile function of the atria and ventricles and the ability of these chambers to adequately fill without excessive pressure determine overall cardiac performance. Thus, abnormalities in systolic or diastolic function may lead to heart failure. Comprehensive knowledge of cardiac anatomy and physiology is crucial for modern anesthesia practice. This chapter describes the fundamentals of cardiac anatomy and physiology, and fluency in this subject is essential for modern anesthesia practice. The authors will focus on the left atria and ventricle (LA and LV, respectively) for the vast majority of the subsequent discussion.
Gross Anatomy
Architecture
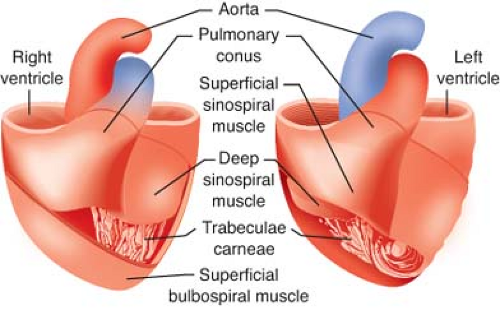 Figure 10.1. Illustration depicting the components of the myocardium. The outer muscle layers pull the apex of the heart toward the base. The inner circumferential layers constrict the lumen, particularly of the LV. (Reproduced with permission from: Barash PG, Cullen BF, Stoelting RK, et al. Clinical Anesthesia, 6th ed., Lippincott Williams & Wilkins, Figure 10-1, p. 210.) |
The LV free walls taper in thickness from base to apex because the relative amount of midmyocardium gradually declines. LV and RV subendocardium and LV midmyocardium extend from the LV anterior wall to form the interventricular septum. As a result, the septum thickens toward the LV chamber during contraction under normal conditions. However, RV distention or chronic pressure-overload may produce paradoxical motion of the interventricular septum. Notably, regional differences in LV wall thickness and fiber orientation contribute to load-dependent alterations in LV mechanics.3 The trabeculae carneae are irregular ridges of subendocardium of unknown significance that are present within the LV apex and, to a greater extent, within the RV chamber.
The LV apex and interventricular septum are relatively fixed in space within the mediastinum, whereas the lateral and posterior walls shift position to the anterior and the right during contraction. This motion effectively changes the LV longitudinal axis from a plane that favors LV filling to a position that facilitates ejection. The systolic movement of the lateral and posterior LV walls also produces the palpable point of maximum impulse. The LV base descends toward the apex during systole as a result of the combined effects of subendocardial and subepicardial contraction, papillary muscle shortening, and recoil caused by ejection of blood into the aortic root. Taken as a whole, synchronous activation of the LV causes its long axis to shorten, reduces its chamber diameter, and produces rotation of its apex in an anterior-right direction (Fig. 10-2). Differential changes in wall tension also create an apex-to-base intraventricular pressure gradient during LV ejection; this effect serves to enhance the transfer efficiency of stroke volume from the LV to the aortic root.
The crescent-shaped RV is anatomically anterior and to the right of the LV. This chamber propels deoxygenated venous blood into the relatively compliant, low-pressure PA tree. As a result, the RV wall is thinner and contains fewer cardiac myocytes compared with the LV, which is subjected to substantially greater wall stress during ejection of blood into the high-pressure systemic vasculature. Embryologically distinct inflow and outflow tracts exist in the RV, which causes RV contraction to be less temporally uniform than the LV. The RV relies on the interventricular septum against which the RV free wall contracts. The LV also contributes to RV contractility. The combination of these factors provides a mechanical advantage to the RV that compensates for its thinner
wall dimension, thereby allowing it to pump a stroke volume that is equal to the LV stroke volume during each beat. Nevertheless, the RV more easily decompensates with acute increases in afterload than the LV because the RV is able to produce less than 20% of the total pressure–volume (stroke) work than the thicker, more muscular LV. However, the RV is highly compliant and is able to accommodate to acute changes in intraventricular volume to a greater degree than the LV.
wall dimension, thereby allowing it to pump a stroke volume that is equal to the LV stroke volume during each beat. Nevertheless, the RV more easily decompensates with acute increases in afterload than the LV because the RV is able to produce less than 20% of the total pressure–volume (stroke) work than the thicker, more muscular LV. However, the RV is highly compliant and is able to accommodate to acute changes in intraventricular volume to a greater degree than the LV.
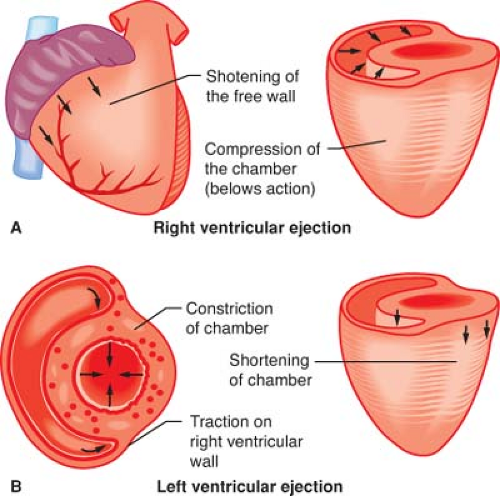 Figure 10.2. Illustration depicting the contraction characteristics and modes of emptying of the RV and LV. The volumes ejected by each ventricle are equal, but the LV requires a more circumferential muscular wall to eject its volume at a pressure that is approximately four to five times greater than that in the right ventricle. (Reproduced with permission from: Barash PG, Cullen BF, Stoelting RK, et al. Clinical Anesthesia, 6th ed., Lippincott Williams and Wilkins, Figure 10-2, p. 210.) |
Valve Structure
Two pairs of translucent, macroscopically avascular valves guarantee unidirectional movement of blood through the heart. The pulmonic and aortic valves separate the RV and LV from the PA and aorta, respectively. These valves open and close and operate passively with changes in pressure resulting from RV and LV contraction and relaxation. The pulmonic valve leaflets are named for their anatomic locations (right, left, and anterior) within the mediastinum, whereas each aortic valve leaflet is identified on the basis of a corresponding coronary artery (e.g., the right coronary leaflet lies adjacent to the ostium of the right coronary artery). At the time of maximal blood flow during ejection, the orifice areas of the pulmonic and aortic valves are nearly equivalent to the corresponding annulus cross-sectional areas. The sinuses of Valsalva are dilated segments in the proximal aortic root that are located immediately superior to each aortic leaflet. Hydraulic flow vortices (eddy currents) occur within the sinuses of Valsalva that prevent adherence of the valve leaflets to the walls of the aortic root during systole and also aid closure of the valve by facilitating leaflet mobility during diastole.4 These actions assure that the aortic valve leaflets do not inadvertently occlude the coronary ostia. Sinuses of Valsalva are absent in the proximal PA.
The mitral valve is located between the LA and the LV, contains two leaflets (the oval-shaped anterior and the crescent-shaped posterior), and resembles a saddle when viewed in three dimensions. Coaptation of the leaflets occurs along a central curve with the posterior leaflet creating its concave border. Despite the differences in their shapes, the anterior and posterior leaflets have similar cross-sectional areas because the posterior leaflet occupies a greater percentage of the annular circumference. Anterior–lateral and posterior–medial commissures connect the leaflets and are located above the corresponding papillary muscles. The mitral valve opens in response to a positive pressure gradient between the LA and LV that occurs during the late phase of relaxation. This process is facilitated by untwisting and elastic recoil of the LV as the chamber relaxes. Closure of the leaflets occurs as retrograde blood flow moves toward the valve during early systole. The chordae tendineae act as “cable stays” to prevent leaflet prolapse and inversion during LV contraction. Primary and secondary chordae tendineae are attached to the leaflet edges and bodies, respectively, whereas tertiary chordae insert into the distal posterior leaflet or the myocardium adjacent to the annulus. The papillary muscles are composed of subendocardial myocardium that contract with the LV. Each papillary muscle has chordal attachments to both mitral leaflets; contraction of the papillary muscles effectively tensions the chordae. As a result, motion of the leaflets beyond the coaptation zone is restricted. Closure of the mitral leaflets is also facilitated by tightening of the annulus because the surrounding subepicardium contracts in a sphincter-like manner. The mitral valve apparatus is critical for overall LV function. The valve apparatus guarantees that blood flow from the LA to the LV is unidirectional because blood is prevented from refluxing into the LA and pulmonary veins during LV contraction. Thus, ischemia or infarction of a papillary muscle may cause dysfunction of the mitral apparatus and is often accompanied by acute mitral regurgitation. In addition, the mitral apparatus plays an essential role in LV systolic function because papillary muscle shortening assists LV apical contraction. This latter action is particularly important during mitral valve replacement because severing attachments between the chordae tendineae and the papillary muscles is invariably associated with a reduction in LV ejection fraction.
The tricuspid valve is composed of anterior, posterior, and septal leaflets, and assures unidirectional movement of blood from the RA and RV. The posterior leaflet is usually smaller than the anterior and septal leaflets. The morphologic RV may be distinguished from the LV by the presence of a septal papillary muscle in patients with transposition of the great vessels and other forms of congenital heart disease. The moderator band is a lateral segment of myocardium stretching between the anterior and septal papillary muscles and separates the embryologic RV inflow and outflow tracts. The RV often has coarser trabeculae carnae than the LV, but the physiologic implications of these differences in subendocardial structure remain a mystery. In contrast to the mitral valve, the tricuspid valve does not have a collagenous annulus. Instead, the tricuspid leaflets originate from the atrioventricular groove that separates the RA from the RV. Notably, the atrioventricular groove also contains the proximal right coronary artery, and this vessel must be carefully avoided during tricuspid valve repair or replacement surgery.
Coronary Blood Supply
The left anterior descending, left circumflex, and right coronary arteries (LAD, LCCA, and RCA, respectively) supply blood to the
LV (Fig. 10-3). Most coronary blood flow to the LV myocardium occurs during diastole because aortic blood pressure is greater than LV pressure. A critical stenosis or acute occlusion of one of these three vessels may produce myocardial ischemia or infarction accompanied by abnormal regional contractile function based on the known distribution of each coronary artery’s blood supply. The medial LV anterior wall, the anterior two-thirds of the interventricular septum, and the LV apex are supplied by the LCA and its branches; the anterior and posterior aspects of the lateral wall are perfused by the LCCA; and the medial portions of the posterior wall and the posterior one-third of the interventricular septum are supplied by the RCA. The “dominance” of the coronary circulation is determined on the basis of which major coronary artery feeds the posterior descending coronary artery (PDA). A “right dominant” circulation occurs when the RCA supplies blood to the PDA and is observed in approximately 80% of patients. A “left dominant” circulation in which the PDA is supplied by the LCCA is observed in the remaining 20% of patients. Distal connections or collateral blood vessels between the major coronary arteries may also provide perfusion to regions of myocardium that lie distal to a severe stenosis or occlusion. A single coronary artery (2:1 ratio of RCA to LCCA) provides blood flow to the posterior–medial papillary muscle in two-thirds of patients. Thus, RCA or LCCA occlusion may produce acute posterior–medial papillary muscle ischemia or infarction and, as a result, new mitral valve dysfunction. However, this is not always the case, as both vessels perfuse the posterior–medial papillary muscle in the remaining patients.5 In contrast to the posterior–medial papillary muscle, the anterior–lateral papillary muscle has a dual blood supply (LAD and LCCA), rendering this papillary muscle less susceptible to ischemic dysfunction than its counterpart.
LV (Fig. 10-3). Most coronary blood flow to the LV myocardium occurs during diastole because aortic blood pressure is greater than LV pressure. A critical stenosis or acute occlusion of one of these three vessels may produce myocardial ischemia or infarction accompanied by abnormal regional contractile function based on the known distribution of each coronary artery’s blood supply. The medial LV anterior wall, the anterior two-thirds of the interventricular septum, and the LV apex are supplied by the LCA and its branches; the anterior and posterior aspects of the lateral wall are perfused by the LCCA; and the medial portions of the posterior wall and the posterior one-third of the interventricular septum are supplied by the RCA. The “dominance” of the coronary circulation is determined on the basis of which major coronary artery feeds the posterior descending coronary artery (PDA). A “right dominant” circulation occurs when the RCA supplies blood to the PDA and is observed in approximately 80% of patients. A “left dominant” circulation in which the PDA is supplied by the LCCA is observed in the remaining 20% of patients. Distal connections or collateral blood vessels between the major coronary arteries may also provide perfusion to regions of myocardium that lie distal to a severe stenosis or occlusion. A single coronary artery (2:1 ratio of RCA to LCCA) provides blood flow to the posterior–medial papillary muscle in two-thirds of patients. Thus, RCA or LCCA occlusion may produce acute posterior–medial papillary muscle ischemia or infarction and, as a result, new mitral valve dysfunction. However, this is not always the case, as both vessels perfuse the posterior–medial papillary muscle in the remaining patients.5 In contrast to the posterior–medial papillary muscle, the anterior–lateral papillary muscle has a dual blood supply (LAD and LCCA), rendering this papillary muscle less susceptible to ischemic dysfunction than its counterpart.
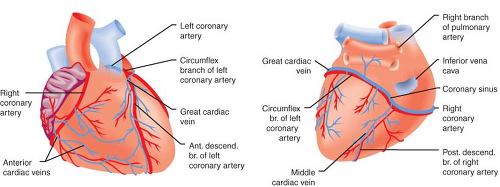 Figure 10.3. An anterior view of the heart (left) shows right coronary and left anterior descending coronary arteries. A posterior view (right) shows left circumflex and posterior descending coronary arteries. The anterior cardiac veins from the RV and the coronary sinus, which drain primarily the LV, empty into the RA. (Reproduced with permission from: Barash PG, Cullen BF, Stoelting RK, et al. Clinical Anesthesia, 6th ed., Lippincott Williams and Wilkins, Figure 10-16, p. 222.) |
the major sources of blood supply to the LA. As a result, LCCA occlusion often causes LA contractile dysfunction, whereas a compensatory increase in LA contractility may be observed if the LAD becomes acutely occluded.6 The RA is supplied by branches of both the RCA and the LCCA. For example, the sinoatrial (SA) node may be perfused by either the RCA (55% of patients) or the LCCA (45%). The atrioventricular (AV) node is more commonly supplied by the RCA, but also may be perfused by the LCCA depending on the right or left dominance of the coronary circulation. The clinical implications of these anatomical relationships are clear: Critical stenosis or acute occlusion of either the RCA or LCCA may interrupt normal conduction and cause bradyarrhythmias.
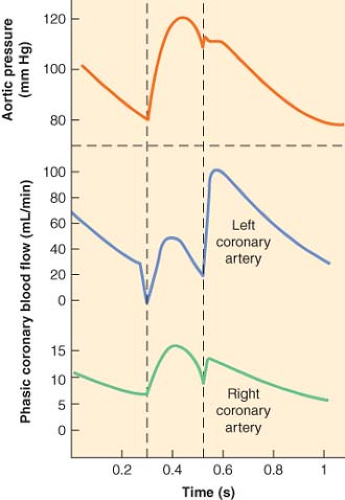 Figure 10.4. Schematic representation of blood flow in the left and right coronary arteries during phases of the cardiac cycle. Note that most left coronary flow occurs during diastole, while right coronary flow (and coronary sinus flow) occurs mostly during late systole and early diastole. (Reproduced with permission from: Barash PG, Cullen BF, Stoelting RK, et al. Clinical Anesthesia, 6th ed., Lippincott Williams and Wilkins, Figure 10-19, p. 224.) |
The proximal branches of the RCA, LCCA, and LAD are located on the epicardial surface of the heart and give rise to multiple intramural vessels that penetrate perpendicularly or obliquely deep into the ventricular walls. Except for the thin tissue layer on the endocardial surface, the heart’s blood supply is almost entirely derived from these three major coronary arteries. The penetrating branches divide into dense capillary networks located roughly along the courses of the myocardial bundles. Arterial branches with diameters between 50 and 500 μm form interconnecting anastomoses throughout the endocardium of the RV and LV walls (Fig. 10-5). Another network of subendocardial vessels between 100 and 200 μm in diameter forms a plexus of deep anastomoses. A coronary collateral circulation may also arise from different branches of the same coronary artery or from branches of two different coronary arteries. Flow through a coronary collateral is usually negligible because the driving pressure at the two ends of the connection is nearly equal. However, if the artery supplying one branch of a collateral vessel becomes severely stenotic or occluded (e.g., atherosclerotic disease), the large pressure reduction will divert blood flow through the patent artery and into the distribution of the occluded artery through the collateral vessel. Thus, the extent of development of the coronary collateral circulation may determine whether myocardial ischemia or infarction is likely in patients with coronary artery disease.
The main coronary venous drainage tends to retrace the course of the major coronary arteries along the AV and interventricular grooves. The great cardiac vein (that runs along the AV groove and the LAD), the anterior cardiac vein (located with the RCA), and the middle cardiac vein (associated with PDA) are the three major coronary veins. In general, there are two coronary veins located along either side of each coronary arterial branch. The coronary veins converge and terminate in the coronary sinus, which empties into the posterior aspect of the right atrium above the tricuspid valve. Approximately 85% of the total coronary blood flow to the LV drains into the coronary sinus. The remaining blood flow empties directly into the atrial and ventricular cavities via the Thebesian veins. The RV veins drain into the anterior cardiac veins; these empty individually into the right atrium.
The structure of the coronary capillary network is similar to that observed in other tissue beds. Because the heart has an exceptionally high metabolic demand, the density of capillary blood vessels to myofibrils in myocardium is approximately 1:1, and adjacent capillaries are typically separated by the diameter of one myocyte. The distribution of capillaries is quite uniform and ranges between 3,000 and 4,000/mm2 of tissue. Notably, capillary density is reduced in the AV node and interventricular septum; this observation may explain why the conducting system is more susceptible to ischemia. As in other capillary beds, coronary capillaries are the sites for exchange of O2 and CO2, and for the movement of larger molecules across the endothelium without the impediment of vascular smooth muscle.
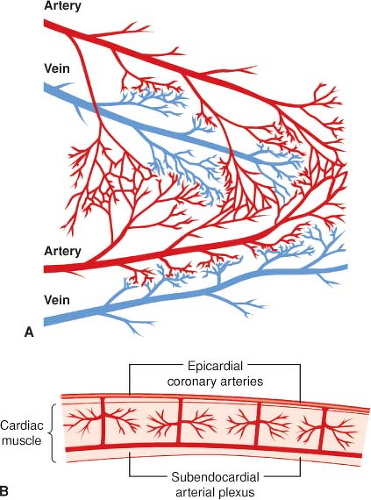 Figure 10-5. A: Diagram of the arterial-to-arterial and venous-to-venous anastomoses of the coronary arterial system, which allows diversion of flow if one distribution becomes blocked. B: Diagram of the epicardial coronary vessels lying on the cardiac muscle surface, the penetrating deep vessels, and the subendocardial arterial plexus connecting the deep vessels. (Reproduced with permission from: Barash PG, Cullen BF, Stoelting RK, et al. Clinical Anesthesia, 6th ed., Lippincott Williams and Wilkins, Figure 10-17, p. 223.) |
Impulse Conduction
How the heart is electrically activated is essential to its mechanical performance. The primary cardiac pacemaker is the SA node, although declines in firing rate, delays or blockade of normal conduction, or the presence of secondary pacemakers (e.g., AV node, bundle of His) may supersede the dominance of SA automaticity. The initial SA node depolarization is rapidly transmitted across the RA to the AV node by the anterior, middle (Wenckebach), and posterior (Thorel) internodal pathways. The SA node depolarization is also transmitted to the LA through the atrial septum by Bachmann’s bundle (a branch of the anterior internodal pathway). It is important to note that examination of myocyte histology rarely differentiates the internodal pathways, although these structures may be clearly identified in the electrophysiology laboratory. The atria are electrically isolated from the ventricles by the heart’s cartilaginous skeleton. As a result, atrial depolarization is directed solely to the RV and the LV through the AV node. Because AV node conduction is relatively slow compared with the pathways proximal and distal to it, the AV node is responsible for the sequential contraction pattern of the atria and the ventricles. Clearly, accessory pathways that bypass the AV node and establish abnormal
conduction between the atria and ventricles may produce supraventricular tachyarrhythmias. This is the putative mechanism by which the bundle of Kent produces Wolff–Parkinson–White syndrome. The AV node transmits its depolarization to the His bundle, which further transmits the signal to the RV and LV via the right and left bundle branches, respectively, through Purkinje fibers within the endocardium. The conduction velocity through the His bundle, the bundle branches, and the Purkinje network is very rapid, assuring coordinated RV and LV depolarization and contraction. In contrast, exogenous cardiac pacing does not rely on the normal conduction sequence and may cause dyssynchronous LV activation, which may be misinterpreted as a new ischemia-induced regional wall motion abnormality during cardiac surgery. Similarly, contractile dyssynchrony produced by chronic RV apical pacing may also lead to LV dysfunction.7 Indeed, cardiac resynchronization therapy in patients with CHF is based on reestablishing the normal sequence of electrical activation because LV contractile synchrony is restored.8
conduction between the atria and ventricles may produce supraventricular tachyarrhythmias. This is the putative mechanism by which the bundle of Kent produces Wolff–Parkinson–White syndrome. The AV node transmits its depolarization to the His bundle, which further transmits the signal to the RV and LV via the right and left bundle branches, respectively, through Purkinje fibers within the endocardium. The conduction velocity through the His bundle, the bundle branches, and the Purkinje network is very rapid, assuring coordinated RV and LV depolarization and contraction. In contrast, exogenous cardiac pacing does not rely on the normal conduction sequence and may cause dyssynchronous LV activation, which may be misinterpreted as a new ischemia-induced regional wall motion abnormality during cardiac surgery. Similarly, contractile dyssynchrony produced by chronic RV apical pacing may also lead to LV dysfunction.7 Indeed, cardiac resynchronization therapy in patients with CHF is based on reestablishing the normal sequence of electrical activation because LV contractile synchrony is restored.8
Coronary Physiology
Blood supply to the LV is directly dependent on the difference between the aortic pressure and LV end-diastolic pressure (coronary perfusion pressure) and inversely related to the vascular resistance to flow, which varies to the fourth power of the radius of the vessel (Poiseuille’s law). Two other determinants of coronary flow are vessel length and viscosity of the blood, but these factors are relatively constant. Resting coronary blood flow in the adult is approximately 250 mL/min (1 mL/min/g) or 5% of total cardiac output. The changes in aortic pressure and the impedance to flow resulting from physical compression of the intramural coronary arteries during the cardiac cycle govern the pulsatile pattern of coronary flow in the LV. The LV subendocardium is exposed to a higher pressure than the subepicardial layer during systole. Indeed, the systolic intraventricular pressure may be higher than the peak LV systolic pressure. Because of these differences in tissue pressure, the subendocardial layer is more susceptible to ischemia in the presence of coronary artery stenoses, pressure-overload hypertrophy, or pronounced tachycardia. Coronary blood flow may also be compromised when aortic diastolic pressure is reduced (e.g., severe aortic insufficiency). Elevated LV end-diastolic pressure, as observed during CHF, also reduces coronary blood flow because of decreased coronary perfusion pressure. Coronary sinus (venous) blood flow is maximal during late systole because of the extravascular compression and the low RA pressure.
The two major determinants of coronary blood flow (perfusion pressure and vascular resistance) also vary substantially during the cardiac cycle. Coronary perfusion pressure certainly changes in response to aortic, intramyocardial, and coronary venous pressures, but the major factor that regulates coronary blood flow is the variable resistance produced by coronary vascular smooth muscle. Sympathetic nervous system activation increases coronary vascular smooth muscle tone. The degree of stretch of smooth muscle (termed the “myogenic factor”) also affects coronary vascular tone. However, metabolic factors are the primary physiologic determinants of coronary vascular tone and, hence, myocardial perfusion. The ratio of epicardial to endocardial blood flow ratio remains near 1.0 throughout the cardiac cycle despite systolic compressive forces exerted on the subendocardium. The more pronounced resistance to flow in the subendocardium is offset by beta-adrenoceptor–mediated vasodilation and by local metabolic autocrine factors (e.g., release of the vasodilator adenosine during hypoxic conditions) produced by the myocardium itself. The relative maintenance of subendocardial blood flow may also be related to the extensive number of redundant arteriolar and capillary anastomoses within the subendocardium.
The heart normally extracts between 75% and 80% of arterial O2 content, by far the greatest O2 extraction of all the body’s organs. The majority of O2 consumption results from the rate and magnitude of LV pressure development during isovolumic contraction. Oxygen consumption is also affected by the diameter of the LV as dictated by the Law of Laplace (see below). Heart rate is the primary determinant of myocardial O2 demand in the intact heart. Increases in myocardial contractility, preload, and afterload are also associated with elevations in myocardial O2 demand. Cardiac O2 extraction is near maximal under resting conditions and cannot be substantially increased during exercise. Thus, the primary mechanism by which myocardium meets its O2 demand is through enhanced O2 delivery, which is proportional to coronary blood flow when hemoglobin concentration is constant. Coronary blood flow and myocardial O2 consumption increase fourfold to fivefold during strenuous physical exercise. The difference between maximal and resting coronary blood flow is termed “coronary reserve.” Myocardial O2 consumption is a major determinant of coronary blood flow. For example, coronary vascular resistance is greater in the rested, perfused heart than in the contracting heart, indicating that coronary blood flow increases in response to a higher rate of O2 consumption. The mechanism(s) responsible for the correlation between myocardial work, O2 consumption, and coronary vessel dilatation has yet to be precisely determined. In addition to metabolically induced vasodilation, the factors responsible for coronary autoregulation (maintenance of coronary blood flow with a change in perfusion pressure) and reactive hyperemia (the severalfold increase in blood flow above baseline after a brief period of ischemia) are also not well understood. Metabolic coronary vasodilation occurs, at least in part, as a result of activation of the sympathetic nerves to the heart and coronary vasculature during an increase in heart rate and myocardial contractility. This sympathetic nerve stimulation produces a feed-forward vasodilation of small coronary arterioles via activation of beta-adrenoceptors.9 An alpha–adrenoceptor-induced vasoconstriction also occurs in larger coronary arteries during exercise. This vasoconstriction occurs upstream from coronary small coronary arterioles and serves two important functions: reduction of vascular compliance and attenuation of coronary blood flow oscillations during the cardiac cycle. These actions act to preserve coronary blood flow to the more vulnerable LV endocardium when heart rate, contractility, and myocardial O2 consumption are elevated. Interestingly, cardiac parasympathetic nerves have a prominent role in regulating heart rate, but these nerves appear to have a negligible direct effect on the regulation of coronary blood flow.
The conclusions about sympathetic nervous system control of the coronary circulation are based on alterations in the slope of the O2 consumption–coronary venous O2 tension relation during graded exercise in the presence of exogenous alpha- or beta-adrenoceptor blockade. Current evidence implicating the beta-adrenoceptor in coronary vasodilation accounts for only about 25% of the total coronary vasodilation observed during exercise-induced hyperemia. These data suggest that the other 75% of coronary vasodilation during exercise may be produced by as yet undefined local metabolic factors that act on coronary vascular smooth muscle with or without the influence of endothelium. Recent evidence indicates that this process may be mediated, at least in part, by the release of adenine nucleotides released from red blood cells that subsequently activate endothelial purinergic receptors to produce vasodilation during exercise.10 Many metabolic factors have been proposed to individually or collectively
modulate coronary flow at the arteriolar or capillary level, including adenosine, nitric oxide, arterial oxygen or CO2 tension, pH, osmolarity, K+, Ca2+, and prostaglandins. Many of these factors exert predictable direct effects. For example, hypoxia or ischemia decreases arterial oxygen tension and pH and increases CO2 tension, adenosine, and K+ and Ca2+ concentrations. These changes may augment coronary blood flow, but none individually appear to be the sole determinant of vasodilation during exercise. For example, adenosine receptor blockade does not alter coronary blood flow under resting conditions or during exercise. Similarly, inhibition of nitric oxide (NO) production or ATP-sensitive K+ (KATP) channels does not alter the slope of the myocardial O2 consumption–coronary venous O2 relationship during graded exercise. Despite these data, it is abundantly clear that NO and KATP channels are important regulators of myocardial O2 supply–demand relations under resting conditions. Adenosine released during hypoxia or ischemia causes coronary vasodilation; this effect is mediated by KATP channel activation. Adenosine and KATP channels have also shown to play a role in reactive hyperemia after ischemia, but these mediators do not appear to be required for coronary autoregulation. Moreover, the KATP channel may act to reduce coronary vascular smooth muscle tone, and thus, maintain a higher basal coronary blood flow, during resting conditions. While not acting as a local metabolic vasodilator, NO may react to increased downstream arterial dilation by dilating larger, upstream epicardial coronary arteries, thereby preventing excessive sheer stress on coronary endothelium.
modulate coronary flow at the arteriolar or capillary level, including adenosine, nitric oxide, arterial oxygen or CO2 tension, pH, osmolarity, K+, Ca2+, and prostaglandins. Many of these factors exert predictable direct effects. For example, hypoxia or ischemia decreases arterial oxygen tension and pH and increases CO2 tension, adenosine, and K+ and Ca2+ concentrations. These changes may augment coronary blood flow, but none individually appear to be the sole determinant of vasodilation during exercise. For example, adenosine receptor blockade does not alter coronary blood flow under resting conditions or during exercise. Similarly, inhibition of nitric oxide (NO) production or ATP-sensitive K+ (KATP) channels does not alter the slope of the myocardial O2 consumption–coronary venous O2 relationship during graded exercise. Despite these data, it is abundantly clear that NO and KATP channels are important regulators of myocardial O2 supply–demand relations under resting conditions. Adenosine released during hypoxia or ischemia causes coronary vasodilation; this effect is mediated by KATP channel activation. Adenosine and KATP channels have also shown to play a role in reactive hyperemia after ischemia, but these mediators do not appear to be required for coronary autoregulation. Moreover, the KATP channel may act to reduce coronary vascular smooth muscle tone, and thus, maintain a higher basal coronary blood flow, during resting conditions. While not acting as a local metabolic vasodilator, NO may react to increased downstream arterial dilation by dilating larger, upstream epicardial coronary arteries, thereby preventing excessive sheer stress on coronary endothelium.
Cardiac Myocyte Anatomy and Function
Ultrastructure
The heart contracts and relaxes nearly 3 billion times during an average lifetime, based on a heart rate of 70 beats per minute and a life expectancy of 75 years. A review of cardiac myocyte ultrastructure provides important insights into how the heart accomplishes this astonishing performance. The sarcolemma is the external membrane of the cardiac muscle cell. The sarcolemma contains ion channels (e.g., Na+, K+, Ca2+), ion pumps and exchangers (e.g., Na+–K+ ATPase, Ca2+-ATPase, Na+–Ca2+ or Na+–H+ exchangers), G-protein–coupled and other receptors (e.g., beta1-adrenergic, adenosine, opioid), and transporter enzymes that regulate intracellular ion concentrations, facilitate signal transduction, and provide metabolic substrates required for energy production. Deep invaginations of the sarcolemma, known as transverse (“T”) tubules, penetrate the internal structure of the myocyte at regular intervals, thereby assuring rapid, uniform transmission of the depolarizing impulses that initiate contraction to be simultaneously distributed throughout the cell. Unlike the skeletal muscle cell, the cardiac myocyte is densely packed with mitochondria, which are responsible for generation of the large quantities of high-energy phosphates (e.g., adenosine triphosphate [ATP]) required for the heart’s phasic cycle of contraction and relaxation. The fundamental contractile unit of cardiac muscle is the sarcomere. The myofilaments within each sarcomere are arranged in parallel cross-striated bundles of thin (containing actin, tropomyosin, and the troponin complex) and thick (primarily composed of myosin and its supporting proteins) fibers. Sarcomeres are connected in series, thereby producing characteristic shortening and thickening of the long and short axes of each myocyte, respectively, during contraction.
Contractile Apparatus
Myosin, actin, tropomyosin, and the three-protein troponin complex compose the six major components of the contractile apparatus. Myosin (molecular weight of approximately 500 kDa; length, 0.17 μm) contains two interwoven chain helices with two globular heads that bind to actin and two additional pairs of light chains. Enzymatic digestion of myosin divides the structure into light (containing the tail section of the complex) and heavy (composed of the globular heads and the light chains) meromyosin. The elongated tail section of the myosin complex functions as the architectural support of the molecule (Fig. 10-6). The globular heads of the myosin dimer contain two “hinges” located at the junction of the distal light chains and the tail helix that play a critical role in myofilament shortening during contraction. These globular structures bind to actin, thereby activating an ATPase that plays a central role in hinge rotation and release of actin during contraction and relaxation, respectively. The maximum velocity of sarcomere shortening has been shown to be dependent on the activity of this actin-activated myosin ATPase. Notably, adult and neonatal atrial and ventricular myocardia contain several different myosin ATPase isoforms that are distinguished by their relative ATPase activity. The myosin molecules are primarily arranged in series along the length of the thick filament, but are abutted “tail-to-tail” in the center of the thick filament. This orientation facilitates shortening of the distance between “Z” lines during contraction as the thin filaments are drawn progressively toward the center of the sarcomere.
The light chains contained within the myosin complex serve either “regulatory” or “essential” roles. Regulatory myosin light
chains may favorably modulate myosin–actin interaction through Ca2+-dependent protein kinase phosphorylation, whereas essential light chains serve an as yet undefined obligate function in myosin activity, as their removal denatures the myosin molecule. Discussion of myosin light chain isoforms is beyond the scope of the current chapter, but isoform switches from ventricular to atrial forms have been observed in left ventricular hypertrophy that may contribute to contractile dysfunction.11 In addition to myosin and its binding protein, thick filaments contain titin, a long elastic protein that attaches myosin to the “Z” lines. Titin has been postulated to be a “length sensor” similar to a bidirectional spring that establishes progressively greater passive restoring forces as sarcomere length approaches its maximum or minimum.12 Compression and stretching of titin occur during decreases and increases in muscle load, thereby resisting further sarcomere shortening and lengthening, respectively. Thus, titin is a third important elastic element (in addition to actin and myosin) that contributes to the stress-strain biomechanical properties of cardiac muscle.13
chains may favorably modulate myosin–actin interaction through Ca2+-dependent protein kinase phosphorylation, whereas essential light chains serve an as yet undefined obligate function in myosin activity, as their removal denatures the myosin molecule. Discussion of myosin light chain isoforms is beyond the scope of the current chapter, but isoform switches from ventricular to atrial forms have been observed in left ventricular hypertrophy that may contribute to contractile dysfunction.11 In addition to myosin and its binding protein, thick filaments contain titin, a long elastic protein that attaches myosin to the “Z” lines. Titin has been postulated to be a “length sensor” similar to a bidirectional spring that establishes progressively greater passive restoring forces as sarcomere length approaches its maximum or minimum.12 Compression and stretching of titin occur during decreases and increases in muscle load, thereby resisting further sarcomere shortening and lengthening, respectively. Thus, titin is a third important elastic element (in addition to actin and myosin) that contributes to the stress-strain biomechanical properties of cardiac muscle.13
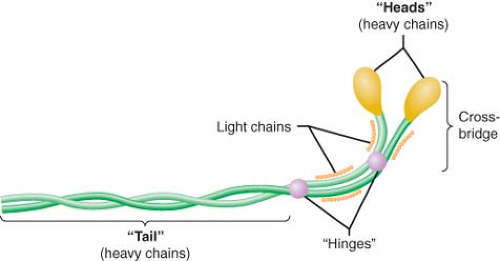 Figure 10.6. Schematic illustration of the myosin molecule demonstrating double helix tail, globular heads that form cross-bridges with actin during contraction, two pairs of light chains, and “hinges” (cleavage sites of proteolytic enzymes) that divide the molecule into meromyosin fragments (see text). (Reproduced with permission from: Barash PG, Cullen BF, Stoelting RK, et al. Clinical Anesthesia, 6th ed., Lippincott Williams and Wilkins, Figure 10-6, p. 214.) |
Actin is the major component of the thin filament. Actin is a 42 kDa, ovoid-shaped, globular protein (“G” form; 5.5 nm in diameter) that exists in a polymerized filamentous (“F”) form in cardiac muscle. F-actin binds adenosine diphosphate (ADP) and a divalent cation (Ca2+ or Mg2+), but unlike myosin, the molecule does not directly hydrolyze high-energy nucleotides such as ATP. F-actin is wound in double-stranded helical chains of G-actin monomers that resemble two intertwined strands of pearls. A single complete helical revolution of filamentous actin is approximately 77 nm in length and contains 14 G-actin monomers. Actin derives its name from its function as the “activator” of myosin ATPase through its reversible binding with myosin. The hydrolysis of ATP by this actin–myosin complex provides the chemical energy required to produce the conformational changes in the myosin heads that drive the cycle of contraction and relaxation within the sarcomere. Tropomyosin is one of two major inhibitors of actin–myosin interaction. Tropomyosin (length of 40 nm; weight between 68 and 72 kDa) is a rigid double-stranded alpha-helix protein linked by a single disulfide bond. Human tropomyosin contains both alpha and beta isoforms (34 and 36 kDa, respectively) and may be present as either a homo- or a heterodimer.14 Tropomyosin stiffens the thin filament through its position within the longitudinal cleft between intertwined F-actin polymers (Fig. 10-7), but its Ca2+-dependent interaction with troponin complex proteins is the mechanism that links sarcolemmal membrane depolarization to actin–myosin interaction in the cardiac myocyte (excitation–contraction coupling). The thin filaments are anchored to “Z” lines by cytoskeletal proteins including alpha- and beta-actinin and nebulette.15
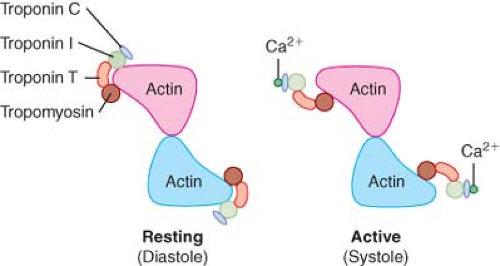 Figure 10.7. Cross-sectional schematic illustration demonstrating the structural relationship between the troponin–tropomyosin complex and actin under resting conditions (left panel) and after Ca2+ binding to troponin C (right panel, see text). (Reproduced with permission from: Barash PG, Cullen BF, Stoelting RK, et al. Clinical Anesthesia, 6th ed., Lippincott Williams and Wilkins, Figure 10-7, p. 215.) |
The troponin proteins serve complementary but distinct roles as critical regulators of the contractile apparatus.16 The troponin complexes are arranged at 40 nm intervals along the length of the thin filament. Troponin C (so named because this molecule binds Ca2+) exists in a highly conserved, single isoform in cardiac muscle. Troponin C is composed of a central nine-turn alpha-helix separating two globular regions that contain four discrete amino acid sequences capable of binding divalent cations including Ca2+ and Mg2+. Of this quartet of amino acid–cation binding sequences, two (termed sites I and II) are Ca2+-specific, thereby allowing the troponin C molecule to respond to the acute changes in intracellular Ca2+ concentration that accompany contraction and relaxation. Troponin I (“inhibitor”) is a 23 kDa protein that exists in a single isoform in cardiac muscle. Troponin I alone weakly prevents the interaction between actin and myosin, but when combined with tropomyosin, the troponin I–tropomyosin complex becomes the major inhibitor of actin–myosin binding. The troponin I molecule contains a serine residue that may be phosphorylated by protein kinase A (PKA) via the intracellular second messenger cAMP, thereby reducing troponin C–Ca2+ binding and enhancing relaxation during administration of beta1-adrenoceptor agonists (e.g., dobutamine) or phosphodiesterase fraction III inhibitors (e.g., milrinone). Troponin T (so denoted because it binds other troponin molecules and tropomyosin) is the largest of the troponin proteins and exists in four major isoforms in human cardiac muscle. Troponin T anchors the other
troponin molecules and may also influence the relative Ca2+ sensitivity of the complex.17
troponin molecules and may also influence the relative Ca2+ sensitivity of the complex.17
Calcium–Myofilament Interaction
Binding of Ca2+ to troponin C precipitates a series of conformational changes in the troponin–tropomyosin complex that lead to the exposure of the myosin-binding site on the actin molecule. During conditions in which intracellular Ca2+ concentration is low (10−7 M; diastole), very little Ca2+ is bound to troponin C, and each tropomyosin molecule is constrained to the outer region of the groove between F-actin filaments by a troponin complex. This structural configuration prevents myosin–actin interaction by effectively blocking cross-bridge formation. Thus, an inhibitory state produced by the troponin–tropomyosin complex exists in cardiac muscle under resting conditions. A 100-fold increase in intracellular Ca2+ concentration (10−5 M; systole) occurs as a consequence of sarcolemmal depolarization, which opens L- and T-type sarcolemmal Ca2+ channels, thereby allowing Ca2+ influx into the myocyte from the extracellular compartment and stimulating Ca2+-dependent Ca2+ release from the SR via its ryanodine receptors. When Ca2+ is bound to troponin C under these conditions, the shape of the troponin C protein becomes elongated and its interactions with troponin I and T are enhanced. These Ca2+-induced allosteric rearrangements in troponin complex structure weaken the interaction between troponin I and actin, allow repositioning of the tropomyosin molecule along the F-actin filaments, and reverse the baseline inhibition of actin–myosin binding by tropomyosin.18 In this way, Ca2+ binding to troponin C may be directly linked to a series of changes in regulatory protein chemical structure that block inhibition of the binding site for myosin on the actin molecule and allow cross-bridge formation and contraction to occur. This antagonism of inhibition is fully reversible, as relaxation is facilitated by dissociation of Ca2+ from troponin C concomitant with rapid restoration of the original conformation of the troponin–tropomyosin complex on F-actin.
Most Ca2+ ions are removed from the myofilaments and the cytosol after membrane repolarization by a Ca2+-ATPase located in the SR membrane (sarcoendoplasmic reticulum Ca2+-ATPase, SERCA). This Ca2+ is stored (concentration of approximately 10−3 M) in the SR bound to calsequestrin and calreticulin until the subsequent sarcolemmal depolarization is initiated. The Na+/Ca2+ exchanger and a Ca2+-ATPase located within the sarcolemmal membrane also remove a small quantity of Ca2+, similar to that which originally entered the myocyte from the extracellular space during depolarization. Phospholamban is a small protein (6 kDa) located in the SR membrane that partially inhibits the activity of the dominant form (type 2a) of cardiac SERCA under baseline conditions. However, phosphorylation of this protein by PKA blocks this inhibition and enhances the rate of SERCA uptake of Ca2+ into the SR,19 thereby increasing the rate and extent of relaxation (positive lusitropic effect) and augmenting the amount of Ca2+ stored for the next cycle of contraction (positive inotropic effect). Thus, SERCA activity is regulated by a cAMP-dependent PKA that is responsive to beta1-adrenoceptor stimulation or phosphodiesterase fraction III inhibition. In addition to PKA-mediated phosphorylation of troponin I that facilitates Ca2+ release from troponin C, these observations explain why positive inotropic drugs such as dobutamine and milrinone also augment relaxation.
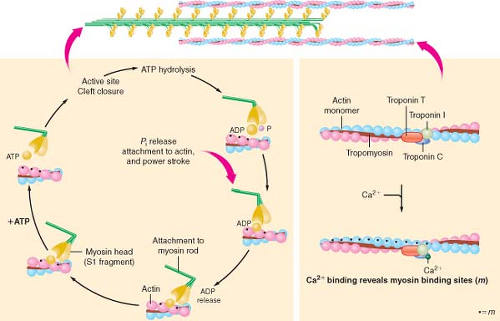 Figure 10.8. Schematic illustration of the actin filaments and its individual monomers and active myosin bindings sites (m; left panel). The myosin head is dissociated from actin by binding with adenosine triphosphate (ATP). Subsequent ATP hydrolysis and release of inorganic phosphate (Pi) “cocks” the head group into a tension-generating configuration. Attachment of the myosin head to actin allows the head to apply tension to the myosin rod and the actin filament. The right panel illustrates Ca2+ binding to troponin C, which causes troponin I to decrease its affinity for actin. As a result, in a conformational shift in tropomyosin position (see text), seven sites on actin monomers are revealed. (Reproduced with permission from: Barash PG, Cullen BF, Stoelting RK, et al. Clinical Anesthesia, 6th ed., Lippincott Williams and Wilkins, Figure 10-9, p. 216.) |
Myosin–Actin Interaction
The biochemistry of cardiac muscle contraction is most often described using a simplified four-component model (Fig. 10-8).20 Binding of ATP with high affinity to the catalytic domain of myosin initiates the series of chemical and mechanical events that cause contraction of the sarcomere to occur. The myosin ATPase enzyme hydrolyzes the ATP molecule into ADP and inorganic phosphate, but these reaction products do not immediately dissociate from myosin. Instead, the ATP hydrolysis products and myosin form an “active” complex that retains the chemical energy released from the reaction as potential energy. In the absence of actin, subsequent dissociation of ADP and phosphate from myosin is the rate-limiting step of myosin ATPase and the muscle remains relaxed. However, the activity of myosin ATPase is markedly accelerated when the myosin–ADP–phosphate complex
is bound to actin, and under these circumstances, the chemical energy obtained from ATP hydrolysis becomes directly transferred into mechanical work. Attachment of myosin to its binding site on the actin molecule releases the phosphate anion from the myosin head, thereby producing a molecular conformation within this cross-bridge structure that generates tension in both myofilaments.21 Release of ADP and the stored potential energy from this activated conformation produce rotation of the cross-bridge (“power stroke”) at the hinge point separating the helix tail region from the globular myosin head and its associated light chain proteins. Each cross-bridge rotation generates 3 to 4 × 10−12 newtons of force and moves myosin approximately 11 nm along the actin molecule.22 Completion of myosin head rotation and ADP release does not dissociate the myosin-active complex but leaves it in a low-energy bound (“rigor”) state. Separation of myosin and actin occurs when a new ATP molecule binds to myosin, and the process is subsequently repeated, provided that energy supply is adequate and the myosin-binding site on actin remains unimpeded by troponin–tropomyosin inhibition.
is bound to actin, and under these circumstances, the chemical energy obtained from ATP hydrolysis becomes directly transferred into mechanical work. Attachment of myosin to its binding site on the actin molecule releases the phosphate anion from the myosin head, thereby producing a molecular conformation within this cross-bridge structure that generates tension in both myofilaments.21 Release of ADP and the stored potential energy from this activated conformation produce rotation of the cross-bridge (“power stroke”) at the hinge point separating the helix tail region from the globular myosin head and its associated light chain proteins. Each cross-bridge rotation generates 3 to 4 × 10−12 newtons of force and moves myosin approximately 11 nm along the actin molecule.22 Completion of myosin head rotation and ADP release does not dissociate the myosin-active complex but leaves it in a low-energy bound (“rigor”) state. Separation of myosin and actin occurs when a new ATP molecule binds to myosin, and the process is subsequently repeated, provided that energy supply is adequate and the myosin-binding site on actin remains unimpeded by troponin–tropomyosin inhibition.
Several factors may affect the efficiency of cross-bridge biochemistry and myocardial contractility independent of autonomic nervous system tone or administration of exogenous vasoactive drugs. There is a direct relationship between myosin ATPase activity and the maximal velocity of unloaded muscle shortening (Vmax). The normal increase in intracellular Ca2+ concentration (from 10−7 to 10−5 M) that occurs after sarcolemmal depolarization enhances baseline myosin ATPase activity fivefold before it interacts with actin, thereby increasing Vmax. Contractile force depends on sarcomere length immediately before sarcolemmal depolarization. This length-dependent activation (Frank–Starling effect) may be related to an increase in myofilament sensitivity to Ca2+, favorable alterations in spacing between myofilaments, or titin-induced elastic recoil. Abrupt increases in load during contraction (Anrep effect) or those that occur after a prolonged pause between beats (Woodworth phenomenon) causes transient increases in contractile force through a length-dependent activation mechanism. An increase in cardiac muscle stimulation frequency also augments contractile force (treppe phenomenon) via enhanced myofilament Ca2+ sensitivity and greater SR Ca2+ release.
Law of Laplace
Related posts:




Full access? Get Clinical Tree


