The pH, defined by Sorenson, is the negative log of the concentration of H+. Therefore, in neutral solution, the pH is 7. This is a very small concentration of [H+], and therefore, addition of small amounts of [H+] to water will lead to significant fluctuations in pH. For example, we will consider an experiment performed by Jorgensen and Astrup (1) in which 1.25 mEq/L of HCl is added to hemolyzed human blood. Assuming that the blood contained no buffers (i.e., if the blood was imagined to be a container of water that starts at a neutral pH), the expected pH following such an infusion would be calculated by the following:
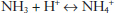
Taking the negative log yields a pH of 1.9; this would be the expected pH if there were not buffers available. However, following the infusion, the pH of the blood changed approximately 0.2 pH points. This means that less than 1/10,000th of the H+ added remains unbound in the blood. This illustrates the tremendous buffering capacity of the blood. A buffer can be thought of as a substance that, when present in solution, takes up [H+] and therefore resists change in pH when [H+] is added. The overall buffering system of the body is complex and includes several components (Table 135.1). The most important system is the carbon dioxide–bicarbonate system, which is the principal buffer in the extracellular fluid (ECF). This buffering system is also very important clinically since it is the only buffering system where the two components (acid and conjugate base) are readily measurable in the ECF. Buffers work by binding the free H+ as the conjugate base, which is a weak acid.
Buffers allow the body to resist acute changes in pH; however, buffering capacity will eventually be depleted if acid is continually added. For example, in humans, the fixed acid production is approximately 70 to 100 mmol/d. It is through the excretion of the daily acid load that ultimately allows the body to maintain acid–base balance. The excretion of the daily acid load occurs through two distinct mechanisms: the renal excretion of fixed acid, and the respiratory excretion of volatile acid (i.e., carbon dioxide). Through the interconversion of bicarbonate, carbonic acid, and carbon dioxide, fixed and volatile acids can be buffered until they can be excreted through the urine or respiration (Fig. 135.1). In the lung, CO2 is released, which ultimately leads to more H+ reacting with HCO3− to generate water and more CO2. In the kidneys, the entire filtered load of bicarbonate is—in order to avoid losing base—reabsorbed. When the kidney excretes one H+ in the urine, one “new” HCO3– is generated. These two processes are both important in the excretion of the acid load, and modulation of these processes is also important in compensating for acid–base disturbances.
Urinary Excretion of Fixed Acids
In the reabsorption of bicarbonate, the corresponding H+ produced in the process must be excreted in the urine. As most bicarbonate reabsorption occurs in the proximal tubule, the renal secretion of H+ is ten times greater in the proximal tubule—approximately 4,000 mmol/d—as compared with the distal tubule—approximately 400 mmol/d. However, in the distal tubule, there is a much higher luminal–intracellular H+ gradient than that seen in the proximal tubule—a ratio of approximately 500:1. This high gradient is due to active secretion of H+ into the tubule. If the excretion of acid occurred in the absence of buffers, the ability to excrete acid in the urine would be limited. In much the same way that the body can absorb large amounts of acid without much change in pH, the kidney accomplishes a similar task in the excretion of large amounts of fixed acid through the use of buffers in the urine with modestly acidic pH (approximately 5.5 under maximal conditions). The excretion of H+ in the urine occurs with different conjugate bases, which are grouped as titratable acids—mostly phosphates—and nontitratable acids—ammonium. The excretion of titratable acid has a limit that is, for the most part, dependent on the filtered load of phosphate, as this is the main buffer for nontitratable acids. However, the kidney can generate its own buffer—ammonia; moreover, the renal capacity to generate ammonia and to excrete acid as ammonium under normal conditions is substantial. This capacity may be significantly upregulated in the face of systemic acidosis. Ammonia is produced in the kidney, traverses the plasma membrane, and is “trapped” in the tubular lumen because the low pH drives the following reaction to the right, as the plasma membrane is much less permeable to the charged species ammonium:

| TABLE 135.1 Blood and Extracellular Fluid Buffers | |
 | |
Therefore, ammonia–ammonium acts as a urinary buffer system, allowing the elimination of one H+ for nearly every ammonia produced. The buffering of acid in the urine, especially via ammonium, allows for substantial amounts of acid to be excreted without generating excessively acidic urine.
Titratable acids make up a relatively small proportion of the acid excreted and do not increase to near the degree that nontitratable acidity (ammonium) increases in the face of systemic acidosis.

FIGURE 135.1 Normal acid–base homeostasis.
The kidney, through active secretion of H+ in the distal tubule, is able to achieve an H+ concentration gradient of approximately 100:1 between the urine and the intracellular space of the tubular epithelial cells. This corresponds to the maximally acidic urine of approximately pH 5. Without any buffers, it would require 7,000 L of urine to excrete a daily load of 70 mEq of acid in buffer-free urine of pH 5.0! Therefore, urinary buffers are very important in allowing the body to excrete the daily fixed acid load. Chronic metabolic acidosis stimulates the renal production of ammonia as a physiologic response; this response reaches its maximum production after several days.
Assessing Urinary Acid Excretion
In the presence of systemic acidosis, the kidney will compensate by increasing the excretion of fixed acids, mainly in the nontitratable form (i.e., ammonium). The increase in ammonium—which is a cation; recall that ammonia is predominantly in the form of NH4+ at a pH of 7 or below—excretion results in a perturbation in the electrolytes present in the urine. This manifests as a change in the urine anion gap where ammonium is the unmeasured anion. The urinary anion gap is a useful clinical test that can be used to gauge the amount of ammonium excreted in the urine without directly measuring it (2). It may be used to indirectly estimate the amount of ammonium in the urine and is calculated using the following formula:

The urine anion gap is assessed in patients with metabolic acidosis and is used to determine if the renal response to the systemic acidosis is appropriate. In other words, the urine anion gap answers the question: Are the kidneys excreting the acid load appropriately or are the kidneys part of the acid–base problem? If the kidneys are excreting the acid load appropriately, there must be a nonrenal source of the acidosis—for example, diarrhea. Because ammonium is not a measured cation in this equation, the presence of significant amounts of ammonium causes an abundance of chloride relative to the measured cationic constituents of the urine (sodium and potassium). Therefore, if there is a high level of ammonium in the urine, the urine anion gap will be negative. The relationship between the amount of ammonium in the urine and the urine anion gap is illustrated in Figure 135.2. As a general rule, a negative urinary anion gap suggests that the kidney is excreting ammonium in the urine. This is a continuous variable, and the more negative the value, the greater the renal response. In the face of systemic acidosis, if the renal response is appropriate, there will be a high amount of ammonium in the urine, and the urine anion gap will be highly negative. This is sometimes referred to as a negative net urinary charge; however, this is a bit misleading because the urine is, of course, electroneutral; it is simply because we are not considering the contribution of ammonium that the net urinary charge seems negative. A highly negative urine anion gap is strong evidence that the renal response to metabolic acidosis is normal. Conversely, an anion gap that is near zero or positive suggests that there is little or no ammonium in the urine, which is reflected by a paucity of chloride in the urine relative to the concentration of measured cations. This is evident that the kidney is not appropriately excreting ammonium in the urine and suggests a renal contribution to the acidosis. A caveat that is often clinically important is that the presence of unmeasured anions in the urine (such as β-hydroxybutyrate) may falsely depress the urinary anion gap, and therefore this test does have some limitations, such as during ketonuria.

FIGURE 135.2 Effect of urine ammonium concentration on the urine anion gap.







Full access? Get Clinical Tree


