29 Biochemical, Cellular, and Molecular Mechanisms of Neuronal Death and Secondary Brain Injury in Critical Care
In this chapter, we provide a general discussion of the biochemical, cellular, and molecular mechanisms of neuronal death and secondary brain injury that are germane to the central nervous system (CNS) insults that require neurointensive care, highlighting the important shared mechanisms in these conditions. In Chapter 30, Dr. Kofke builds upon the biochemical and molecular mechanisms to address general pathophysiologic principles in neurointensive care, focusing on intracranial dynamics and the cerebral circulation. Chapters 31 through 42 of Part 2 address other important facets of neurointensive care, such as monitoring and coma, along with the specific pathophysiology and treatment of the key disease processes central to neurointensive care in both adults and children. This includes traumatic brain injury (TBI), cardiopulmonary arrest, stroke, subarachnoid hemorrhage (SAH), and seizures, among other insults.
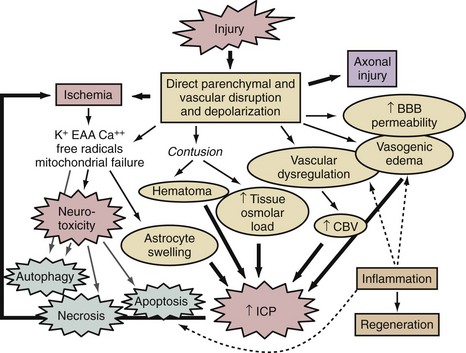
A thumbnail sketch of the most important mechanisms of secondary injury involved in the brain after a traumatic or ischemic insult is provided in Figure 29-1. Central to all brain insults relevant to neurointensive care is the occurrence of cerebral ischemia and/or cerebral energy failure. The principal consequence of ischemic injury and/or energy failure is neuronal death. The two principal forms of ischemia in neurointensive care are global and focal, as seen in cases of cardiopulmonary arrest and stroke, respectively.
 Global Cerebral Ischemia
Global Cerebral Ischemia
In patients with global cerebral ischemia, insults are dense and often square-wave in nature.1 The classic example of a global cerebral ischemic insult in neurointensive care is ventricular fibrillation cardiopulmonary arrest (see Chapter 33). Using conventional approaches, patients can be successfully resuscitated from these insults only if they are brief—that is, circulation must be restored in 5 to 12 minutes, although the maximal duration compatible with intact neurologic outcome can depend on a variety of factors, such as temperature. In cases of complete global cerebral ischemia, adenosine triphosphate (ATP) and phosphocreatine levels in brain are depleted in less than 2 minutes.2,3 Membrane failure ensues, with loss of ion homeostasis that includes cellular release of K+ and uptake of Ca++, Na+, and Cl−.2,3 Upon reperfusion, a complex sequence of events is set into motion that depends on the duration of the insult. Disturbances in lipid metabolism such as free fatty acid release and DNA damage result, along with a series of deleterious cascades including oxidative and nitrosative stress, excitotoxicity, poly-ADP-ribose polymerase (PARP) activation, mitochondrial and endoplasmic reticulum (ER) dysfunction, and a host of cell-signaling abnormalities. A number of endogenous neuroprotectant responses are also initiated. The specific biochemical, cellular, and molecular events are discussed later. The aforementioned increases in intracellular calcium level are believed to play a critical role in initiating many of these events. In situations where the patient is potentially salvageable, such as with threshold insults, reperfusion results in transient hyperemia (minutes) followed by delayed hypoperfusion (hours).1,4,5 The pattern of neuronal damage seen after global cerebral ischemia is classically termed selective vulnerability. This is often delayed and primarily neuronal in nature, and it is believed to result from complex biological cascades involving some features of programmed cell death (discussed later).
A number of brain regions are specifically vulnerable to ischemia, including the CA1 region of the hippocampus, cortical layers 3 and 5, portions of the amygdaloid nucleus, and cerebellar Purkinje cells, among others.2,3,5 Global ischemic insults from cardiopulmonary arrest from which there is some potential for recovery are generally believed to be devoid of important increases in intracranial pressure, since, based on studies in animal models, it has been shown that the threshold for producing poor outcome in patients with global ischemic insults is less than that needed to generate clinically significant intracranial hypertension.6 Thus, brain edema and vascular injury are not believed to represent important therapeutic targets after global cerebral ischemia. Two relevant but atypical global insults in neurointensive care are asphyxial cardiopulmonary arrest (particularly important in children and discussed in Chapter 42), and near-hanging episodes. In the latter, obstruction of cerebral venous drainage during the asphyxial insult compounds the ischemic insult.
 Focal Cerebral Ischemia
Focal Cerebral Ischemia
Focal ischemic insults in neurointensive care are produced by thrombotic or embolic events and generally produce a dense ischemic focus that is surrounded by a periischemic penumbral region with intermediate cerebral blood flow (CBF) values.2 The ischemic focus is generally believed to be unsalvageable unless reperfused almost immediately. In contrast, the ischemic penumbra is a region with some collateral flow and represents a therapeutic target for reperfusion with thrombolytics and/or pharmacologic therapy. In cases of focal cerebral ischemia, a hierarchy of CBF thresholds has been demonstrated in experimental studies, with inhibition of protein synthesis being the most sensitive to CBF reductions, followed by loss of electrical activity (evoked potentials and electroencephalogram), and eventually membrane failure.7–8 Unlike the selective vulnerability seen in global ischemic insults, focal cerebral ischemia produces pan-necrosis of the vasculature and astrocytes, resulting in infarction. However, cell death in the penumbra can demonstrate necrotic, apoptotic, and mixed phenotypes. Again, however, classic apoptosis is not seen. Astrocyte swelling and blood-brain barrier injury with focal cerebral edema can play important roles. In the penumbra, spreading depression waves resulting in depolarization can enhance excitotoxic damage with expansion of the lesion core. Reperfusion can occur spontaneously or with the administration of thrombolytics and can produce a microcosm of the aforementioned oxidative and nitrosative stress, mitochondrial and ER damage, and cell signaling abnormalities seen in global cerebral ischemia. In patients with focal cerebral ischemia with large infarcts, brain swelling can be substantial enough that secondary ischemia can result from intracranial hypertension. Dr. Kofke discusses these concepts in greater detail in Chapter 30. Focal cerebral ischemia from delayed vasospasm is also the most common critical complication of SAH and is discussed in Chapter 35.
 Traumatic Brain Injury
Traumatic Brain Injury
In cases of severe TBI, the biochemical and molecular mechanisms involved depend on the specific type of injury. In cases of focal contusion, direct disruption of parenchyma with local necrosis and hemorrhage results in superimposed vascular disruption, blood-brain barrier permeability, and local ischemia. This sets the stage for excitotoxicity and necrotizing cascades in the contusion penumbra, including oxidative and nitrosative stress, and calpain-mediated proteolysis, among other mechanisms.9,10 Local axonal injury is also seen in patients with contusions. Focal contusions are commonly complicated by marked local swelling and often by intracranial hypertension, with the potential for secondary focal or global ischemic insults or herniation syndromes. In contrast in diffuse injury, a constellation of diffuse axonal and vascular disruption can be seen, with characteristic findings of petechial hemorrhages in the white matter.11 This insult can be devastating even in the absence of intracranial hypertension.12 The biochemical and molecular events involved in axonal injury are discussed later. In cases of severe TBI, combined insults that include both multiple contusions and diffuse injury are also common. Finally, in addition to secondary compression ischemia from refractory intracranial hypertension, secondary extracerebral insults such as hypotension and hypoxemia can also negatively affect outcome and, importantly, complicate the biochemical and molecular response to severe TBI, markedly enhancing delayed neuronal death in brain regions that might otherwise have recovered.13,14
 Key Biochemical and Molecular Mechanisms of Neuronal Secondary Damage
Key Biochemical and Molecular Mechanisms of Neuronal Secondary Damage
Excitotoxicity
Excitotoxicity describes the process by which glutamate and other excitatory amino acids cause neuronal damage. Lucas and Newhouse15 first described the toxicity of glutamate. Olney16 subsequently reported that intraperitoneal administration of glutamate produces brain injury in experimental animals. Although glutamate is the most abundant neurotransmitter in the brain, exposure to toxic levels produces neuronal death.17 Glutamate exposure produces neuronal injury in two phases. Minutes after exposure, sodium-dependent neuronal swelling occurs.18 This is followed by delayed calcium-dependent degeneration. These effects are mediated through both ionophore-linked receptors, labeled according to specific agonists (N-methyl-D-aspartate [NMDA], kainite, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]), and receptors linked to second messenger systems, called metabotropic receptors. Activation of these receptors leads to calcium influx through receptor-gated or voltage-gated channels, or through the release of intracellular calcium stores. Increased intracellular calcium concentration is the trigger for a number of processes that can lead to cellular injury or death (Figure 29-2). One mechanism involves activation of neuronal nitric oxide synthase (nNOS), leading to nitric oxide (NO) production, peroxynitrite formation, and resultant DNA damage. PARP is an enzyme normally operative in DNA repair. In the face of overwhelming DNA damage, PARP overactivation leads to depletion of NAD+ and ATP, metabolic failure, and cell death.19–21 PARP may also impair ATP production directly via posttranslational modification of electron transport chain proteins.22 This may be important, since PARP knockout mice exhibit improved outcome versus controls after experimental stroke or TBI.20,23
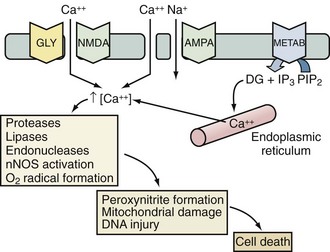
There is considerable evidence in experimental laboratory models supporting an important contribution of excitotoxicity to the evolution of secondary damage in cases of global and focal cerebral ischemia, severe TBI, SAH, and status epilepticus.24–31 Evidence supporting an important role for excitotoxicity in humans has similarly been provided in cases of severe TBI, stroke, and SAH. Persson and Hillered32 reported increases in brain interstitial levels of glutamate in a patient with SAH as early as 1992. Palmer et al.33 first demonstrated increased concentrations of excitatory amino acids in ventricular cerebrospinal fluid (CSF) from adult patients with TBI. Glutamate concentrations were about fivefold greater than in control patients (up to 7 µM)—levels sufficient to cause neuronal death in cell culture.34 Bullock et al.35 characterized patterns of glutamate release by measuring excitatory amino acids by microdialysis in patients after TBI. Patients with a normal head computed tomography (CT) scan and no secondary ischemic events had interstitial concentrations of glutamate that were increased early in their course then returned to normal. In contrast, patients with a progressively rising level of glutamate died. Similarly, in cases of human stroke, Bullock et al.36 also reported massive increases in the excitatory amino acids, glutamate and aspartate, in a patient who required decompressive craniectomy to prevent brainstem herniation.
Despite these and many other clinical reports, clinical trials with anti-excitotoxic therapies have been unsuccessful in patients with either stroke or TBI. This may be due to problems with patient selection, side effects of the anti-excitotoxic agents that were tested, and the likelihood that treatment was initiated too late.37 Inhibition of plasticity by anti-excitotoxic therapies may also limit their efficacy, especially at the interface between the acute and subacute periods after injury.38
Programmed Cell Death Cascades
It is now increasingly clear from experimental models and human data that cells dying after global or focal cerebral ischemia or TBI can be categorized on a morphologic continuum ranging from necrosis to apoptosis.39,40 Recently, additional phenotypic definitions have been included within this continuum: those of autophagic degeneration, programmed necrosis, and “parthanatos.”41–43 Apoptosis is a morphologic description of cell death defined by cell shrinkage and nuclear condensation, internucleosomal DNA fragmentation, and the formation of apoptotic bodies.44 In contrast, cells dying of necrosis display cellular and nuclear swelling with dissolution of membranes. Apoptosis requires a cascade of intracellular events for completion of cell death; thus, the term apoptosis was previously used synonymously with programmed cell death.45 Because other types of cell death have now been characterized that can also be considered “programmed,” apoptosis now refers primarily to the phenotypic definition as classically defined by Kerr et al.44 In diseases with complex and multiple mechanisms, such as stroke and TBI, it is typically the rule rather than the exception to detect dying cells with many or all currently defined cell-death phenotypes.46 For example, some cells may display DNA fragmentation and activation of proteases involved in apoptosis, despite having nuclear and cellular swelling. Dying cells with mixed phenotypes may represent particularly difficult therapeutic targets.
Biochemical Pathways in Delayed Neuronal Death
Apoptosis is an evolutionarily conserved process required for selective cell elimination during development, and it occurs in all tissues, including brain. Execution of apoptosis requires novel gene expression and protein synthesis.47–49 Apoptosis is an intricate and critical mechanism for balancing cell proliferation, remodeling of tissues during development, and maintenance of tissues with a high rate of cell turnover. Apoptosis can be thought of as “molecular débridement,” delicately eliminating unwanted cells, with minimal disturbance of neighboring cells. Apoptosis is cybernetic and may occur via multiple pathways that can be independent (Figure 29-3); however, cross-talk between these (and other nonapoptotic) pathways also may occur.50,51 At present, neuronal apoptosis can be segregated into two pathways, one involving the activation of a family of cysteine proteases termed caspases, and one that is caspase independent.52
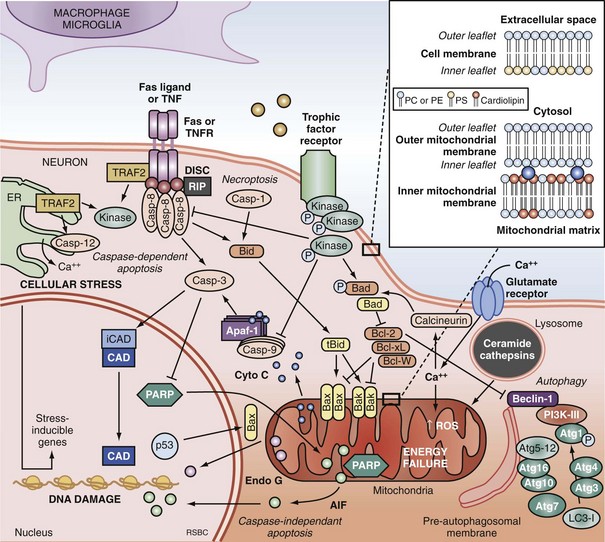
Caspase family proteases include 14 currently identified members that are synthesized as proenzymes, which for the most part are proteolytically activated.50 Initiator caspases, including caspase 8, 9, and 10, are activated by autocleavage and aggregation. Executioner caspases, including caspase 3, 6, and 7, are cleaved and activated by initiator caspases. The proteolytic cleavage of caspase substrates produces the phenotypic changes characteristic of apoptosis, including cytoskeletal disintegration, DNA fragmentation, and disruption of cellular and DNA repair processes (Figure 29-4). Cytoskeletal caspase targets include spectrin and nuclear lamin53; in addition, caspase 3 activates the enzyme, gelsolin, which cleaves actin.54 Active caspase 3 can also cleave the inhibitor of caspase-dependent deoxyribonuclease, permitting caspase-dependent deoxyribonuclease to digest DNA into small oligonucleosomal fragments.55 These small DNA fragments (multiples of approximately 180 base pairs) can be seen on a DNA gel as a ladder and are a hallmark of caspase-dependent apoptosis. Caspase 3 also inhibits DNA repair by proteolytically inactivating many DNA repair proteins, including PARP.56–58 This combination of features—silencing of the genome and incapacitation of DNA repair processes, and destruction of key cytoskeletal components, all with surgical-like precision and ultimately leading to cell death—illustrates why apoptosis has been referred to as “cell suicide.”
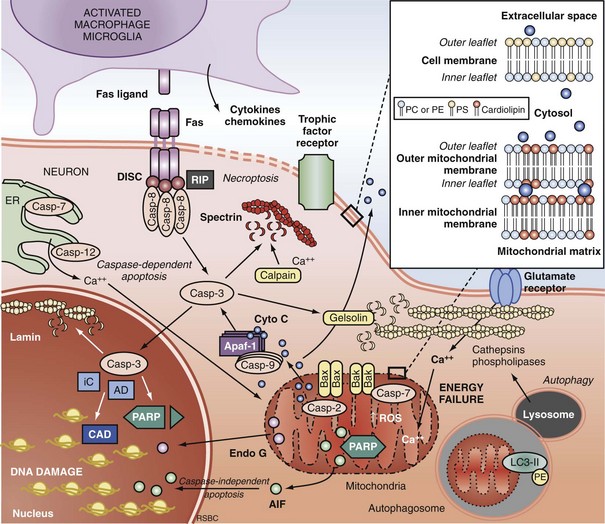
Extrinsic Pathways of Apoptosis and Programmed Necrosis
Programmed cell death can be initiated by extrinsic or intrinsic signals. Extrinsic signals include cell surface death receptor-ligand interactions and cell signaling pathways. The most prominent cell death receptor family is the tumor necrosis factor (TNF) receptor superfamily, which includes TNF-α and Fas.59 The coupling of cell surface TNF or Fas receptors with extracellular TNF-α or Fas ligand induces trimerization of the receptors that leads to the formation of submembrane complexes with intracellular death domain–signaling molecules. This death-inducing signaling complex then activates caspase 860 or 10.61 Caspase 3 is then cleaved and activated, perpetuating the cascade. The extrinsic pathway can also be regulated by multiple intracellular signal transduction pathways that are initiated by G-protein coupled cell surface receptors, which can be either activated by neurotransmitters (e.g., cyclic nucleotides) or inactivated by interruption of trophic factors (e.g., nerve growth factor) after injury.62 Perturbations in neurotransmitters and trophic factors controlling these pathways occur after ischemia and TBI. Multiple interrelated pro-death or pro-survival kinase pathways have been identified, including those involving mitogen-activated protein kinases, and protein kinase B and protein kinase C.63,64 Caspase 8 and 3 cleavage consistent with activation has been demonstrated in humans after TBI.39,65
More recently, programmed cell death with phenotypic characteristics of necrosis (programmed necrosis) or shared characteristics of apoptosis and necrosis (necroptosis) have been described.66 Programmed necrosis occurs through TNF receptor signaling involving receptor interacting protein 1 (RIP-1) and TNF receptor-associated factors (TRAFs) and regulation by protein ubiquitination and phosphorylation.66 Effector mechanisms of programmed necrosis are thought to involve caspase 8 but may also occur via direct effects on mitochondrial permeability transition. Thus, caspase 8 activation may reflect either extrinsic apoptosis or programmed necrosis, or both.
Intrinsic Pathways of Apoptosis
The intrinsic pathway of apoptosis is triggered by stress on cellular organelles, notably mitochondria and ER. Mitochondrial stress can lead to caspase-dependent apoptosis via mitochondrial release of cytochrome C induced upon mitochondrial membrane depolarization. Egress of cytochrome C into the cytosol enables interaction with apoptotic protease activating factor-1 (Apaf-1), dATP, and procaspase 9 to form a complex termed an apoptosome. Apaf-1 activates caspase 9 and subsequently caspase 3.67 Several mitochondrial proteins are capable of inducing apoptosis without direct activation of the caspase cascade, thus exemplifying pathways that are caspase independent. Apoptosis-inducing factor (AIF) within the mitochondria serves as an antioxidant68; however, upon mitochondrial membrane depolarization, it can translocate from the mitochondria to the nucleus, where it is sufficient to induce apoptosis.69 Translocation of AIF into the nuclei induces the formation of large-scale DNA fragmentation (>50 kilobase pairs), in contrast to cytochrome C-mediated, caspase-dependent apoptosis, which leads to oligonucleosomal DNA fragmentation (180-1200 base pairs). AIF-mediated apoptosis occurs in neurons under conditions of experimental TBI52 and cerebral ischemia.70 It is now accepted that PARP-1 overactivation mediates AIF-translocation and subsequent cell death.19,71 As noted earlier, “parthanatos” was recently coined to describe poly(ADP-ribose)-related cell death (from the Greek thanatos, referring to the personification of death).42
Other mitochondrial proteins related to programmed cell death include endonuclease G,72 Htr2A/Omi,73 and Smac/Diablo74; however, their roles in neuronal death after brain injury remain unexplored. Disruption of ER calcium homeostasis and/or accumulation of excess proteins can lead to ER stress, which in turn can trigger programmed cell death via activation of ER-localized caspase 12, an upstream initiator caspase. ER stress-related activation of caspase 12 has been detected in experimental models of cerebral ischemia75 and TBI.76
Autophagic Neurodegeneration
Autophagy is a homeostatic physiologic process important for recycling amino acids by digestion of proteins and organelles. Literally meaning “eating oneself,” this is an important response to nutrient deprivation in every organism. Like apoptosis, disrupted autophagy results in disease, in this case resulting in accumulation of intracellular proteins and aged organelles.77 Possibly like apoptosis, too much autophagy may also contribute to disease, depending upon the insult, organ, and cell type involved. For example, even under conditions of starvation, inhibition of autophagy protects neurons, whereas it exacerbates cell death in fibroblasts.78 Although there is considerable controversy regarding its role, increased autophagy has been demonstrated in models of cerebral ischemia79 and TBI80 and in brain tissue from humans with critical illness.81 The controversy arises in terms of whether or not inhibition or promotion of autophagy is beneficial after brain injury, insofar as both of these divergent strategies have been shown to be protective in various experimental models.80,82–84 There is also cross-talk between autophagy and apoptosis, perhaps at the level of the Bcl-2 protein family.51,85
Regulation of Programmed Cell Death by the Bcl-2 Protein Family
Caspase-dependent and caspase-independent apoptosis, as well as autophagy, are regulated by the B-cell lymphoma-2 (Bcl-2) family of proteins. The Bcl-2 family contains both pro-death and pro-survival members.86 Bcl-2 family proteins regulate changes in permeability of the mitochondrial outer membrane independent of permeability transition pore formation. Bcl-2 family proteins contain highly conserved Bcl-2 homology domains (BH1-BH4) essential for homo- and hetero-complex formation.87 Complexes formed between proteins containing BH3 domains such as Bax, truncated Bid, and Bad can facilitate mitochondrial cytochrome C release.88,89 The antiapoptotic members Bcl-2, Bcl-xL, and Mcl-1L prevent the release of mitochondrial proteins by inhibiting the pore formation.90 Bax expression is associated with neuronal cell death after cardiac arrest in dogs.91 Transgenic mice overexpressing Bcl-2 are partially protected from the neuropathologic sequelae of TBI versus wild-type mice,92 and overexpression of Bcl-xL also inhibits neuronal cell death after focal cerebral ischemia.93 The Bcl-2 interacting partner Beclin 1 contains a BH3-only domain and is required for autophagy.94 It is postulated that binding of Beclin 1 to Bcl-2 or Bcl-xL via the BH3 domain is how cross-talk occurs between apoptosis and autophagy.85
Programmed Cell Death in Human Brain Injury
Phenotypic descriptions of programmed cell death occurring after brain injury in humans date back to the 1940s.95,96 However, biochemical evidence of programmed cell death after brain injury in humans has been reported only within the last decade and has now been reported after TBI,39,65,81,97,98 stroke,99 and epilepsy.100 Brain tissue samples from TBI patients requiring decompressive craniectomy for the treatment of life-threatening intracranial hypertension were found to have evidence of DNA fragmentation by terminal deoxynucleotidyl transferase–mediated nick-end labeling (TUNEL) and cleavage of caspase 1 and 3, suggesting activation of the apoptotic cascade.39 The up-regulation of caspase-8 in human brain after TBI at both transcriptional and translational levels has also been reported.65 Caspase 8 was found predominantly in neurons and was associated with relative levels of the death receptor Fas, providing evidence of the extrinsic apoptotic pathway within neurons. Increases in Fas and Fas ligand have also been reported in CSF from TBI patients, with Fas levels correlating with intracranial pressure.101,102 Activation of the intrinsic apoptotic pathway also occurs after TBI. Alteration of Bcl-2-family proteins has been reported in human brain from adults and in CSF from infants and children after TBI.39,97,103 In pediatric patients, lower concentrations of Bcl-2 were detected in patients who died than in those who survived, supporting a pro-survival role for Bcl-2.97 After TBI in adults, the presence of pro-death Bcl-2 family protein Bax in patients in whom Bcl-2 was also detectable represented a more favorable outcome as compared with patients in whom Bax but not Bcl-2 was detectable.104 In contrast to TBI patients, patients after stroke demonstrate reductions in soluble Bcl-2 and soluble Fas within CSF,99 suggesting dysregulation of apoptosis after stroke. In adolescents and young adults with refractory seizures, increases in Bcl-2 and Bcl-xL, as well as increases in expression and proteolysis of caspase 1 and 3, occur in resected temporal lobe.100 These patients have had medically refractory seizures for several years, implying both protracted and acute apoptosis within the brain. Protracted programmed cell death after TBI also occurs. Cells with apoptotic morphologies and DNA damage detected by TUNEL have been reported in autopsy specimens from patients dying up to 12 months after injury,105 perhaps implying that a relatively wide therapeutic window exists for the administration of treatments aimed at reducing programmed cell death.
Many of these clinical observational studies suggest potential sex differences in cell death mechanisms operative after brain injury. For example, CSF levels of cytochrome C are associated with female gender after TBI in children,106 and CSF levels of the biochemical footprint of PARP activation are associated with male gender after TBI in both children107 and adults.108 These studies are strikingly consistent with experimental studies of neuronal death in vitro109 and in vivo.110,111
Several notes of caution are in order. First, it is unclear what the quantitative contribution of programmed cell death, particularly apoptosis, is in clinical cases of cerebral ischemia or TBI. It is likely that dying cells demonstrate some biochemical and phenotypic features of programmed cell death, but that the actual deathblow to the cell is not dependent on an active process.112 Even if programmed cell death mechanisms do play a key role, it is unclear whether inhibiting neuronal death after injury is entirely beneficial, since apoptosis is a vital mechanism for biological systems to eliminate abnormal or aging cells, and autophagy is important for protein and organelle turnover. In other words, quiet elimination via “cell suicide” of damaged or dysfunctional cells and/or organelles may lead to overall benefit to the patient, in essence “molecular débridement.” Only clinical trials of novel therapies targeting individual programmed cell death cascades will be able to determine whether these mechanisms, alone or in combination, represent important targets in neurointensive care. Recent studies of the efficacy of mild hypothermia after experimental and clinical cardiopulmonary arrest, however, suggest that the success of this intervention may be derived from its effects on programmed cell death.113–115



Full access? Get Clinical Tree


