Autonomic Nervous System: Physiology and Pharmacology
Loreta Grecu
Key Points
Related Matter
Catecholamine Chemical Structures
Renin Angiotensin
Antimuscarinics
Reversible Anticholinesterases
Anesthesia and the Autonomic Nervous System
Anesthesiology is the practice of autonomic medicine. Drugs that produce anesthesia may also have potent autonomic side effects. The greater part of our training and practice is spent acquiring skills in utilizing or averting the autonomic nervous system (ANS) side effects of anesthetic drugs under a variety of pathophysiologic conditions. The success of any anesthetic depends upon how well homeostasis is maintained. The anesthetic chart reflects ANS function.
Table 15-1. Homeostatic Balance Between Adrenergic and Cholinergic Effects | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Afferent fibers from visceral structures are the first link in the reflex arcs of the ANS, and may relay visceral pain or changes in vessel stretch. Most ANS efferent fibers are accompanied by sensory fibers that are now commonly recognized as components of the ANS. However, the afferent components of the ANS cannot be as distinctively divided as can the efferent nerves. ANS visceral sensory nerves are anatomically indistinguishable from somatic sensory nerves. The clinical importance of visceral afferent fibers is closely implicated in the management of chronic pain states.
Functional Anatomy
The ANS falls into two divisions by anatomy, physiology, and pharmacology. Langley divided this nervous system into two parts in 1921. He retained the term “sympathetic” (sympathetic nervous system, SNS) introduced by Willis in 1665 for the first part, and introduced the term “parasympathetic” (parasympathetic nervous system, PNS) for the second. The term ANS was adopted as a comprehensive name for both. Table 15-1 lists the complementary
effects of SNS (adrenergic, sympathetic) and PNS (cholinergic, parasympathetic) activity of organ systems.
effects of SNS (adrenergic, sympathetic) and PNS (cholinergic, parasympathetic) activity of organ systems.
Central Autonomic Organization
Pure central ANS versus somatic centers are not known. Integration of ANS activity occurs at all levels of the cerebrospinal axis. Efferent ANS activity can be initiated locally and by centers located in the spinal cord, brain stem, and hypothalamus. The cerebral cortex is the highest level of ANS integration. Fainting at the sight of blood is an example of this higher level of somatic and ANS integration. ANS function has also been successfully modulated through conscious, intentional efforts demonstrating that somatic responses are always accompanied by visceral responses and vice versa.
The principal site of ANS organization is the hypothalamus. SNS functions are controlled by nuclei in the posterolateral hypothalamus. Stimulation of these nuclei results in a massive discharge of the sympathoadrenal system. PNS functions are governed by nuclei in the midline and some anterior nuclei of the hypothalamus. The anterior hypothalamus is involved with regulation of temperature. The supraoptic hypothalamic nuclei regulate water metabolism and are anatomically and functionally associated with the posterior lobe of the pituitary (see Interaction of Autonomic Nervous System Receptors). This hypothalamic–neurohypophyseal connection represents a central ANS mechanism that affects the kidney by means of antidiuretic hormone. Long-term blood pressure control, reactions to physical and emotional stress, sleep, and sexual reflexes are regulated through the hypothalamus.
The medulla oblongata and pons are vital centers of acute ANS organization. Together, they integrate momentary hemodynamic adjustments and maintain the sequence and automaticity of ventilation. Integration of afferent and efferent ANS impulses at this central nervous system (CNS) level is responsible for the tonic activity exhibited by the ANS. Tonicity holds visceral organs in a state of intermediate activity that can either be diminished or augmented by altering the rate of nerve firing. The nucleus tractus solitarius, located within the medulla, is the primary area for relay of afferent chemoreceptor and baroreceptor information from the glossopharyngeal and vagus nerves. Increased afferent impulses from these two nerves inhibits peripheral SNS vascular tone, producing vasodilation; it also increases vagal tone, producing bradycardia. Studies of patients with high spinal cord lesions show that a number of reflex changes are mediated at the spinal or segmental level. ANS hyperreflexia is an example of spinal cord mediation of ANS reflexes without integration of function from higher inhibitory centers.1
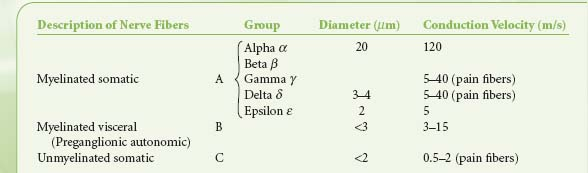
Table 15-2. Classification of Nerve Fibers | |
|---|---|
|
Peripheral Autonomic Nervous System Organization
The peripheral ANS is the efferent (motor) component of the ANS and consists of the same two complementary parts: The SNS and the PNS. Most organs receive fibers from both divisions (Fig. 15-1). In general, activities of the two systems produce opposite but complementary effects (see Table 15-1). A few tissues, such as sweat glands and spleen, are innervated by only SNS fibers. Although the anatomy of the somatic and ANS sensory pathways is identical, the motor pathways are characteristically different. The efferent somatic motor system, like somatic afferents, is composed of a single (unipolar) neuron with its cell body in the ventral gray matter of the spinal cord. Its myelinated axon extends directly to the voluntary striated muscle unit. In contrast, the efferent (motor) ANS is a two-neuron (bipolar) chain from the CNS to the effector organ. The first neuron of both the SNS and PNS originates within the CNS but does not make direct contact with the effector organ. Instead, it relays the impulse to a second station known as an ANS ganglion, which contains the cell body of the second ANS (postganglionic) neuron. Its axon contacts the effector organ. Thus, the motor pathways of both divisions of the ANS are schematically a serial two-neuron chain consisting of a preganglionic neuron and a postganglionic effector neuron (Fig. 15-2).
Preganglionic fibers of both subdivisions are myelinated with diameters of less than 3 mm.1 Impulses are conducted at a speed of 3 to 15 m/s. The postganglionic fibers are unmyelinated and conduct impulses at slower speeds of less than 2 m/s. They are similar to unmyelinated visceral and somatic afferent C fibers (Table 15-2). Compared with the myelinated somatic nerves, the ANS conducts impulses at speeds that preclude its participation in the immediate phase of a somatic response.
Sympathetic Nervous System
The efferent SNS is referred to as the thoracolumbar nervous system. Figure 15-1 demonstrates the distribution of the SNS and its innervation of visceral organs. The preganglionic fibers of the SNS (thoracolumbar division) originate in the intermediolateral gray column of the 12 thoracic (T1–12) and the first three lumbar segments (L1–3) of the spinal cord. The myelinated axons of these nerve cells leave the spinal cord with the motor fibers to form the white (myelinated) communicating rami (Fig. 15-3). The rami enter one of the paired 22 sympathetic ganglia at their respective segmental levels. Upon entering the paravertebral ganglia of the lateral sympathetic chain, the preganglionic fiber may follow one of three courses: (1) synapse with postganglionic fibers in
ganglia at the level of exit, (2) course upward or downward in the trunk of the SNS chain to synapse in ganglia at other levels, or (3) track for variable distances through the sympathetic chain and exit without synapsing to terminate in an outlying, unpaired, SNS collateral ganglion (Fig. 15-3). The adrenal gland is an exception to the rule. Preganglionic fibers pass directly into the adrenal medulla without synapsing in a ganglion (Fig. 15-2). The cells of the medulla are derived from neuronal tissue and are analogous to postganglionic neurons.
ganglia at the level of exit, (2) course upward or downward in the trunk of the SNS chain to synapse in ganglia at other levels, or (3) track for variable distances through the sympathetic chain and exit without synapsing to terminate in an outlying, unpaired, SNS collateral ganglion (Fig. 15-3). The adrenal gland is an exception to the rule. Preganglionic fibers pass directly into the adrenal medulla without synapsing in a ganglion (Fig. 15-2). The cells of the medulla are derived from neuronal tissue and are analogous to postganglionic neurons.
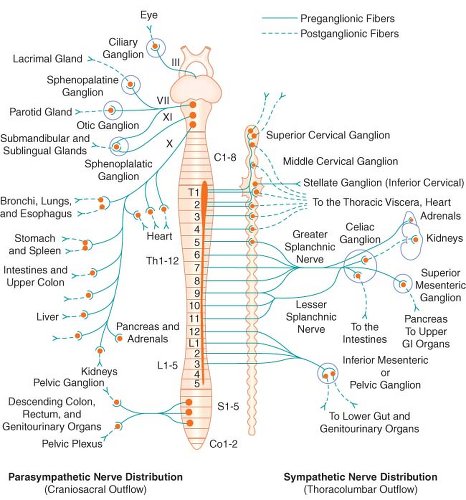 Figure 15.1. Schematic distribution of the craniosacral (parasympathetic) and thoracolumbar (sympathetic) nervous systems. Parasympathetic preganglionic fibers pass directly to the organ that is innervated. Their postganglionic cell bodies are situated near or within the innervated viscera. This limited distribution of parasympathetic postganglionic fibers is consistent with the discrete and limited effect of parasympathetic function. The postganglionic sympathetic neurons originate in either the paired sympathetic ganglia or one of the unpaired collateral plexuses. One preganglionic fiber influences many postganglionic neurons. Activation of the SNS produces a more diffuse physiologic response rather than a discrete, localized effect. GI, gastrointestinal. |
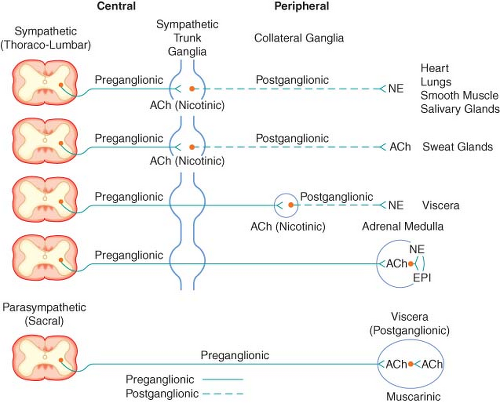 Figure 15.2. Schematic diagram of the efferent ANS. Efferent impulses are integrated centrally and sent reflexly to the adrenergic and cholinergic receptors. Sympathetic fibers ending in the adrenal medulla are preganglionic, and acetylcholine (ACh) is the neurotransmitter. Stimulation of the chromaffin cells, acting as postganglionic neurons, releases epinephrine (EPI) and norepinephrine (NE). |
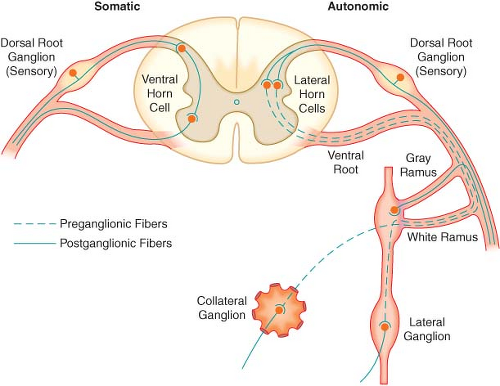 Figure 15.3. The spinal reflex arc of the somatic nerves is shown on the left. The different arrangements of neurons in the sympathetic system are shown on the right. Preganglionic fibers coming out through white rami may make synaptic connections following one of three courses: (1) synapse in ganglia at the level of exit, (2) course up or down the sympathetic chain to synapse at another level, or (3) exit the chain without synapsing to an outlying collateral ganglion. |
The sympathetic postganglionic neuronal cell bodies are located in ganglia of the paired lateral SNS chain or unpaired collateral ganglia in more peripheral plexuses. Collateral ganglia, such as the celiac and inferior mesenteric ganglia (plexus), are formed by the convergence of preganglionic fibers with many postganglionic neuronal bodies. SNS ganglia are almost always located closer to the spinal cord than to the organs they innervate. The sympathetic postganglionic neurons can therefore originate in either the paired lateral paravertebral SNS ganglia or one of the unpaired collateral plexus. The unmyelinated postganglionic fibers then proceed from the ganglia to terminate within the organs they innervate. Many of the postganglionic fibers pass from the lateral SNS chain back into the spinal nerves, forming the gray (unmyelinated) communicating rami at all levels of the spinal cord (Fig. 15-2). They are distributed distally to sweat glands, pilomotor muscle, and blood vessels of the skin and muscle. These nerves are unmyelinated C type fibers (Table 15-2) and are carried within the somatic nerves. Approximately 8% of the fibers in the average somatic nerve are sympathetic.
The first four or five thoracic spinal segments generate preganglionic fibers that ascend in the neck to form three special paired ganglia. These are the superior cervical, middle cervical, and cervicothoracic ganglia. The last is known as the stellate ganglion and is actually formed by the fusion of the inferior cervical and first thoracic SNS ganglia. These ganglia provide sympathetic innervation of the head, neck, upper extremities, heart, and lungs. Afferent pain fibers also travel with these nerves, accounting for chest, neck, or upper extremity pain with myocardial ischemia.
Activation of the SNS produces a diffuse physiologic response (mass reflex) rather than discrete effects. SNS postganglionic neurons outnumber the preganglionic neurons in an average ratio of 20:1 to 30:1.2 One preganglionic fiber influences a larger number of postganglionic neurons, which are dispersed to many organs.
Parasympathetic Nervous System
The PNS, like the SNS, has both preganglionic and postganglionic neurons. The preganglionic cell bodies originate in the brain stem and sacral segments of the spinal cord. PNS preganglionic fibers are found in cranial nerves III (oculomotor), VII (facial), IX (glossopharyngeal), and X (vagus). The sacral outflow originates in the intermediolateral gray horns of the second, third, and fourth sacral nerves. Figure 15-1 shows the distribution of the PNS division and its innervation of visceral organs.
The vagus (cranial nerve X) nerve has the most extensive distribution of all the PNS, accounting for more than 75% of PNS activity. The paired vagus nerves supply PNS innervation to the heart, lungs, esophagus, stomach, small intestine, proximal half of the colon, liver, gallbladder, pancreas, and upper portions of the ureters. The sacral fibers form the pelvic visceral nerves, or nervi erigentes. These nerves supply the remainder of the viscera that are not innervated by the vagus. They supply the descending colon, rectum, uterus, bladder, and lower portions of the ureters and are primarily concerned with emptying. Various sexual reactions are also governed by the sacral PNS. The PNS is responsible for penile erection, but SNS stimulation governs ejaculation.
In contrast to the SNS division, PNS preganglionic fibers pass directly to the organ that is innervated. The postganglionic cell bodies are situated near or within the innervated viscera
and generally are not visible. The proximity of PNS ganglia to or within the viscera provides a limited distribution of postganglionic fibers. The ratio of postganglionic to preganglionic fibers in many organs appears to be 1:1 to 3:1 compared with the 20:1 found in the SNS system. Auerbach’s plexus in the distal colon is the exception, with a ratio of 8,000:1. The fact that PNS preganglionic fibers synapse with only a few postganglionic neurons is consistent with the discrete and limited effect of PNS function. For example, vagal bradycardia can occur without a concomitant change in intestinal motility or salivation. Mass reflex action is not a characteristic of the PNS. The effects of organ response to PNS stimulation are outlined in Table 15-1.
and generally are not visible. The proximity of PNS ganglia to or within the viscera provides a limited distribution of postganglionic fibers. The ratio of postganglionic to preganglionic fibers in many organs appears to be 1:1 to 3:1 compared with the 20:1 found in the SNS system. Auerbach’s plexus in the distal colon is the exception, with a ratio of 8,000:1. The fact that PNS preganglionic fibers synapse with only a few postganglionic neurons is consistent with the discrete and limited effect of PNS function. For example, vagal bradycardia can occur without a concomitant change in intestinal motility or salivation. Mass reflex action is not a characteristic of the PNS. The effects of organ response to PNS stimulation are outlined in Table 15-1.
Autonomic Innervation
Heart
The physiologic importance of the PNS on myocardial contractility is not as well understood as that of the SNS. Cholinergic blockade can double the heart rate (HR) without altering contractility of the left ventricle. Vagal stimulation of the heart can reduce left ventricular maximum rate of tension development (dP/dT) and decrease contractile force by as much as 10% to 20%. However, PNS stimulation is relatively unimportant in this regard compared with its predominant effect on HR. The SNS has the same supraventricular distribution as the PNS, but with stronger representation to the ventricles. SNS efferents to the myocardium funnel through the paired stellate ganglia. The right stellate ganglion distributes primarily to the anterior epicardial surface and the interventricular septum. Right stellate stimulation decreases systolic duration and increases HR. The left stellate ganglion supplies the posterior and lateral surfaces of both ventricles. Left stellate stimulation increases mean arterial pressure and left ventricular contractility without causing a substantial change in HR. Normal SNS tone maintains contractility approximately 20% above that in the absence of any SNS stimulation.4 Therefore, the dominant effect of the ANS on myocardial contractility is mediated primarily through the SNS. Intrinsic mechanisms of the myocardium, however, can maintain circulation quite well without the ANS, as evidenced by the success of cardiac transplants (see Chapter 51 Transplant anesthesia). The heart and ANS are in perfect symbiosis. ANS via its components imprints the cardiac electrophysiology by potentially inducing significant dysrhythmias or electrocardiographic abnormalities, which in the end may lead to global cardiac dysfunction. The precise role of the ANS is unknown, specifically if it is an active component or just an accompaniment. Future research interests concern the modification of the autonomic cardiac innervation through pharmacology or using alternative approaches.5 Early investigations, performed in anesthetized, open-chest animals, demonstrated that cardiac ANS nerves exert only slight effects on the coronary vascular bed; however, more recent studies on chronically instrumented, intact, conscious animals show considerable evidence for a strong SNS regulation of the small coronary resistance and larger conductance vessels6,7 (see below Adrenergic Receptors).
Different segments of the coronary arterial tree react differently to various stimuli and drugs. Normally, the large conductance vessels contribute little to overall coronary vascular resistance (see Chapter 10 Cardiac Anatomy and Physiology). Fluctuations in resistance reflect changes in lumen size of the small, precapillary vessels. Blood flow through the resistance vessels is regulated primarily by the local metabolic requirements of the myocardium. The larger conductance vessels, however, can constrict markedly due to neurogenic stimulation. Neurogenic influence also assumes a greater role in the resistance vessels when they become hypoxic and lose autoregulation.
Peripheral Circulation
Basal vasomotor tone is maintained by impulses from the lateral portion of the vasomotor center in the medulla oblongata that continually transmits impulses through the SNS, maintaining partial arteriolar and venular constriction. Circulating epinephrine (EPI) from the adrenal medulla has additive effects. This basal ANS tone maintains arteriolar constriction at an intermediate diameter. The arteriole, therefore, has the potential for either further constriction or dilation. If the basal tones were not present, the SNS could only affect vasoconstriction and not vasodilation.8 The SNS tone in the venules produces little resistance to flow compared with the arterioles and the arteries. The importance of SNS stimulation of veins is to reduce or increase their capacity. By functioning as a reservoir for approximately 80% of the total blood volume, small changes in venous capacitance produce large changes in venous return and, thus, cardiac preload.
Lungs
The lungs are innervated by both the SNS and PNS. Postganglionic SNS fibers from the upper thoracic ganglia (stellate) pass to the lungs to innervate the smooth muscles of the bronchi and pulmonary blood vessels. PNS innervation of these structures is via the vagus nerve. SNS stimulation produces bronchodilation and pulmonary vasoconstriction.9 Little else has been proven conclusively about the vasomotor control of the pulmonary vessels other than that they adjust to accommodate the output of the right ventricle. The effect of stimulation of the pulmonary SNS nerves on pulmonary vascular resistance is not ideal but may be important in maintaining hemodynamic stability during stress and exercise by balancing right and left ventricular output. Stimulation of the vagus nerve produces almost no vasodilation of the
pulmonary circulation. Hypoxic pulmonary vasoconstriction is a local phenomenon capable of providing a faster adjustment to the organism needs.
pulmonary circulation. Hypoxic pulmonary vasoconstriction is a local phenomenon capable of providing a faster adjustment to the organism needs.
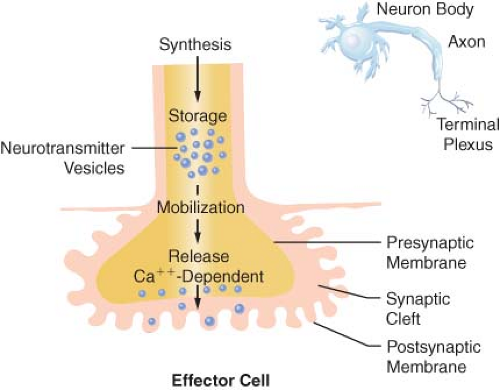 Figure 15.4. The anatomy and physiology of the terminal postganglionic fibers of sympathetic and parasympathetic fibers are similar. |
Both the SNS and the vagus nerve provide active bronchomotor control. SNS stimulation causes bronchodilation whereas vagal stimulation produces constriction. PNS stimulation may also increase secretions of the bronchial glands. Vagal receptor endings in the alveolar ducts also play an important role in the reflex regulation of the ventilation cycle. The lung has important non-ventilatory activity as well. It serves as a metabolic organ that removes local mediators such as norepinephrine (NE) from the circulation and converts others, such as angiotensin 1, to active compounds10 (see below Interaction with Other Regulatory Systems).
Autonomic Nervous System Transmission
Transmission of excitation across the terminal junctional sites (synaptic clefts) of the peripheral ANS occurs through the mediation of released chemicals (Fig. 15-4). Transmitters interact with receptors on the end organ to evoke a biologic response.
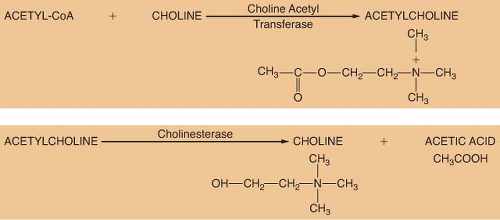 Figure 15.5. Synthesis and metabolism of acetylcholine. |
The terminations of the postganglionic fibers of both ANS subdivisions are anatomically and physiologically similar. The terminations are characterized by multiple branchings called terminal effector plexuses, or reticulae. These filaments surround the elements of the effector unit “like a mesh stocking”.8 Thus, one SNS postganglionic neuron, for example, can innervate ∼25,000 effector cells, for example, vascular smooth muscle. The terminal filaments end in presynaptic enlargements called varicosities. Each varicosity contains vesicles, ∼500 μm in diameter, in which the neurotransmitters are stored (Fig. 15-4). The rate of synthesis depends on the level of ANS activity and is regulated by local feedback. The distance between the varicosity and the effector cell (synaptic or junctional cleft) varies from 100 μm in ganglia and arterioles to as much as 20,000 μm in large arteries. The time for diffusion is directly proportional to the width of the synaptic gap. Depolarization on the nerve releases the vesicular contents into the synaptic cleft by exocytosis.
Parasympathetic Nervous System Transmission
Synthesis
ACh is considered the primary neurotransmitter of the PNS. ACh is formed in the presynaptic terminal by acetylation of choline with acetyl coenzyme A. This step is catalyzed by choline acetyl transferase (Fig. 15-5). ACh is then stored in a concentrated form in presynaptic vesicles. A continual release of small amounts of ACh, called quanta, occurs during the resting state. Each quantum results in small changes in the electrical potential of the synaptic end plate without producing depolarization. These are known as miniature end-plate potentials. Arrival of an action potential causes a synchronous release of hundreds of quanta, resulting in depolarization of the end plate. Release of ACh from the vesicles is dependent on influx of calcium (Ca2+) from the interstitial space. ACh is not reused like NE; therefore, it must be synthesized constantly.
Metabolism
The ability of a receptor to modulate function of an effector organ is dependent upon rapid recovery to its baseline state after stimulation. For this to occur, the neurotransmitter must be quickly removed from the vicinity of the receptor. ACh removal occurs by rapid hydrolysis by acetylcholinesterase (Fig. 15-5). This enzyme is found in neurons, at the neuromuscular junction, and in various other tissues of the body. A similar enzyme, pseudocholinesterase or plasma cholinesterase is also found throughout the body but only to a limited extent in neural tissue. It does not appear to be physiologically important in termination of the action of ACh. Both acetylcholinesterase and pseudocholinesterase hydrolyze ACh as well as other esters (such as the ester-type local anesthetics), and they may be distinguished by specific biochemical tests.3
Sympathetic Nervous System Transmission
Traditionally, the catecholamines EPI and NE are considered the main mediators of peripheral SNS activity. NE is released from localized presynaptic vesicles of nearly all postganglionic sympathetic nerves. Vascular SNS nerve terminals, however, also release ATP. Thus, ATP and NE are co-neurotransmitters. They are released directly to there site of action. Their postjunctional effects appear to be synergistic in tissues.
The SNS fibers ending in the adrenal medulla are preganglionic, and ACh is the neurotransmitter (see Fig. 15-2). It interacts with the chromaffin cells in the adrenal medulla, causing release of EPI and NE. The chromaffin cells take the place of the postganglionic neurons. Stimulation of the sympathetic nerves innervating the adrenal medulla, however, causes the release of large quantities of a mixture of EPI and NE into the circulation. The greater portion of this hormonal surge is normally EPI. EPI and NE, when released into the circulation, are classified as hormones in that they are synthesized, stored, and released from the adrenal medulla to act at distant sites.
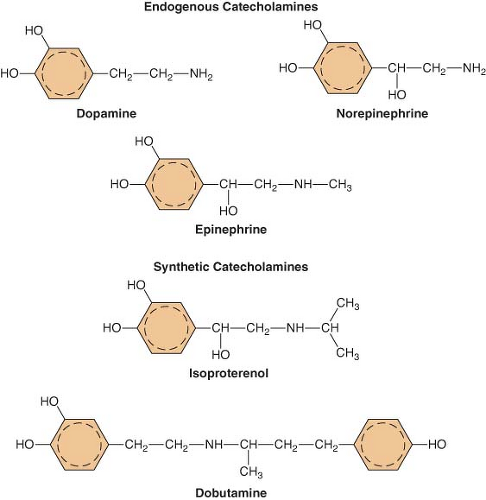 Figure 15.6. The chemical configurations of three endogenous catecholamines are compared with those of three synthetic catecholamines. Sympathomimetic drugs differ in their hemodynamic effects largely because of differences in substitution of the amine group on the catechol nucleus. |
Hormonal EPI and NE have essentially the same effects on effector cells as those caused by local direct sympathetic stimulation. The hormonal effects, although brief, last about 10 times as long as those caused by direct stimulation. EPI has a greater metabolic effect than NE. It can increase the metabolic rate of the body as much as 100%. It also increases glycogenolysis in the liver and muscle with glucose release into the blood. These functions are all necessary to prepare the body for fight or flight.
Catecholamines: The First Messenger
but circulating catecholamines do not cross the blood–brain barrier. The catecholamines present in the brain are synthesized there.
Catecholamines are often referred to as adrenergic drugs because their effector actions are mediated through receptors specific for the SNS. Sympathomimetics can activate these same receptors because of their structural similarity. For example, clonidine is an α2-receptor agonist that does not possess a catechol nucleus and even has two ring systems that are aplanar to each other. However, clonidine enjoys a remarkable spatial similarity to NE that allows it to activate the α receptor. Drugs that produce sympathetic-like effects but lack basic catecholamine structure are defined as sympathomimetics. All clinically useful catecholamines are sympathomimetics, but not all sympathomimetics are catecholamines. The effects of endogenous or synthetic catecholamines on adrenergic receptors can be direct or indirect. Indirect-acting catecholamines (i.e., ephedrine) have little intrinsic effect on adrenergic receptors but produce their effects by stimulating release of the stored neurotransmitter from SNS nerve terminals. Some synthetic and endogenous catecholamines stimulate adrenergic receptor sites directly (e.g., phenylephirine), whereas others have a mixed mode of action. The actions of direct-acting catecholamines are independent of endogenous NE stores; however, the indirect-acting catecholamines are entirely dependent on adequate neuronal stores of endogenous NE.
Synthesis
The main site of NE synthesis is in or near the postganglionic nerve endings. Some synthesis does occur in vesicles near the cell body that pass to the nerve endings. Phenylalanine or tyrosine is taken up into the axoplasm of the nerve terminal and modified into either NE or EPI. Figure 15-7 demonstrates this synthesis cascade. Tyrosine hydroxylase catalyzes the conversion of tyrosine to dihydroxyphenylalanine. This is the rate-limiting step at which NE synthesis is controlled through feedback inhibition. Dopamine (DA) synthesis occurs in the cytoplasm of the neuron. The vesicles of peripheral postganglionic neurons contain the enzyme dopamine-b-hydroxylase, which converts dopamine to NE. The adrenal medulla additionally contains phenylethanolamine-N-methyltransferase, which converts NE to EPI. This reaction takes place outside the medullary vesicles, and the newly formed EPI then enters the vesicle for storage (Fig. 15-8). All the endogenous catecholamines are stored in presynaptic vesicles and released on arrival of an action potential. Excitation–secretion coupling in sympathetic neurons is Ca2+-dependent.
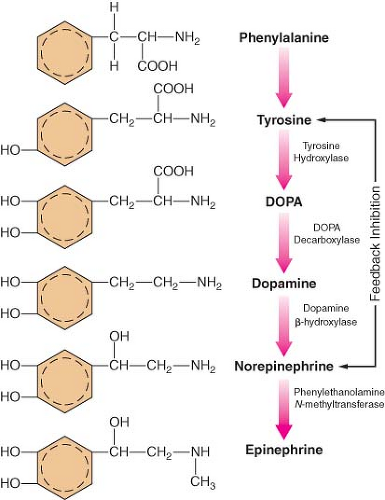 Figure 15.7. Schematic of the synthesis of catecholamines. The conversion of tyrosine to DOPA by tyrosine hydroxylase is inhibited by increased NE synthesis. Epinephrine is shown in these steps but is primarily synthesized in the adrenal medulla. DOPA, dihydroxyphenylalanine. |
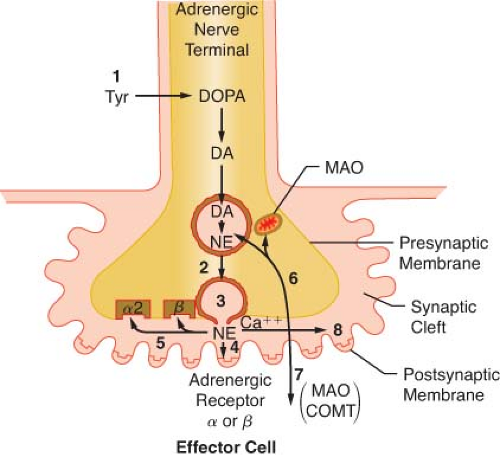 Figure 15.8. Schematic of the synthesis and disposition of NE in adrenergic neurotransmission. (1) Synthesis and storage in neuronal vesicles; (2) Action potential permits calcium entry with (3) exocytosis of NE into synaptic gap. (4) Released NE reacts with receptors on effector cell. NE (5) may react with presynaptic α-2 receptor to inhibit further NE release or with presynaptic β-receptor to enhance reuptake of NE (6) (uptake 1). Extraneuronal uptake (uptake 2) absorbs NE into effector cell (7) with overflow occurring systemically (8). MAO, monoamine oxidase; COMT, catechol-O-methyltransferase; Tyr, tyrosine; DOPA, dihydroxyphenylalanine; NE, norepinephrine. |
Regulation
Increased SNS nervous activity, as in congestive heart failure or chronic stress, stimulates the synthesis of catecholamines. Glucocorticoids from the adrenal cortex stimulate an increase in phenylethanolamine-N-methyltransferase that methylates NE to EPI.
The release of NE is dependent upon depolarization of the nerve and an increase in calcium ion permeability. This release is inhibited by colchicine and prostaglandin E2, suggesting a contractile mechanism. NE inhibits its own release by stimulating presynaptic (prejunctional) α2 receptors. Phenoxybenzamine and phentolamine, α-receptor antagonists, increase the release of NE by blocking inhibitory presynaptic α2 receptors (Fig. 15-9). Other receptors are also important in NE regulation (see below Other Receptors).
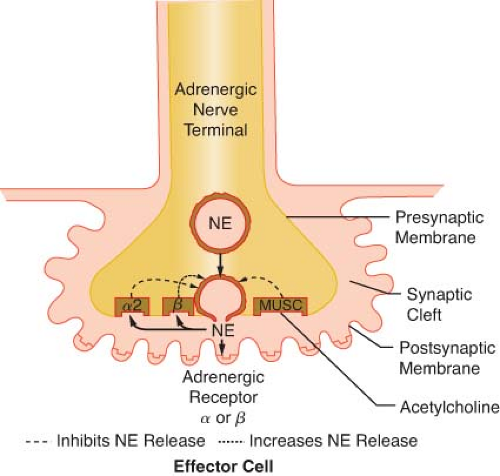 Figure 15.9. This schematic demonstrates just a few of the presynaptic adrenergic receptors thought to exist. Agonist and antagonist drugs are clinically available for these receptors (see Table 15-5). The α-2 receptors serve as a negative feedback mechanism whereby NE stimulation inhibits its own release. Presynaptic β-stimulation increases NE uptake, augmenting its availability. Presynaptic muscarinic (MUSC) receptors respond to ACh diffusing from nearby cholinergic terminals. They inhibit NE release and can be blocked by atropine. NE, norepinephrine. |
Inactivation
The catecholamines are removed from the synaptic cleft by three mechanisms (Fig. 15-8). These are reuptake into the presynaptic terminals, extraneuronal uptake, and diffusion. Termination of NE action at the effector site is almost entirely by reuptake of NE into the terminals of the presynaptic neuron. This is an active, energy-requiring, and temperature-dependent process. The reuptake of NE in the presynaptic terminals is also a stereospecific process. Structurally similar compounds (guanethidine, metaraminol) may enter the vesicles and displace the neurotransmitter. Tricyclic antidepressants and cocaine inhibit the reuptake of NE, resulting in high synaptic NE concentrations and accentuated receptor response. In addition, evidence suggests that NE reuptake is mediated by a presynaptic β-adrenergic mechanism because β-blockade causes marked elevations of EPI and NE11 (see Figs. 15-8 and 15-9). Extraneuronal uptake is a minor pathway for inactivating NE. Effector cells and other extraneuronal tissues take up NE. The NE that is taken up by the extraneuronal tissue is metabolized by monoamine oxidase (MAO) and by catechol-O-methyltransferase (COMT) to form vanillylmandelic acid. The minute amount of catecholamine that escapes these two mechanisms diffuses into the circulation, where it is metabolized by the liver and kidney. The same enzymes inactivate EPI. Reuptake is the predominant pathway for inactivation of the endogenous catecholamines, while metabolism by the liver and kidney is the predominant pathway for catecholamines given exogenously. This accounts for the longer duration of action of the exogenous catecholamines than that noted at the local synapse.
The final metabolic product of the catecholamines is vanillylmandelic acid. Vanillylmandelic acid constitutes the major metabolite (80% to 90%) of NE found in the urine. Less than 5% of released NE appears unchanged in the urine. The metabolic products excreted in the urine provide a gross estimate of SNS activity and can facilitate the clinical diagnosis of pheochromocytoma (see Chapter 46, Endocrine Function).
Receptors
Cholinergic Receptors
ACh is the neurotransmitter for three distinct classes of receptors. These receptors can be differentiated by their anatomic location and their affinity for various agonists and antagonists. ACh mediates the “first messenger” function of transmitting impulses within the PNS, the ganglia of the SNS, and the neuroeffector junction of striated, voluntary muscle (see Fig. 15-2). Cholinergic receptors are further subdivided into muscarinic and nicotinic receptors because muscarine and nicotine stimulate them selectively. However, both muscarinic and nicotinic receptors respond to ACh (see below Cholinergic Drugs). Muscarine activates cholinergic receptors at the postganglionic PNS junctions of cardiac and smooth muscle. Muscarinic stimulation is characterized by bradycardia, decreased inotropism, bronchoconstriction, miosis, salivation, gastrointestinal hypermotility, and increased gastric acid secretion (see Table 15-1). Muscarinic receptors can be blocked by atropine without effect on nicotinic receptors (see below Cholinergic Drugs). Muscarinic receptors are known to exist in sites other than PNS postganglionic junctions. They are found on the presynaptic membrane of sympathetic nerve terminals in the myocardium, coronary vessels, and peripheral vasculature (Fig. 15-9). These are referred to as adrenergic muscarinic receptors because of their location; however, ACh stimulates them also. Stimulation of these receptors inhibits release of NE in a manner similar to α2-receptor stimulation. Muscarinic blockade removes the inhibition of NE release, augmenting SNS activity. Atropine, the prototypical muscarinic blocker, may produce sympathomimetic activity in this manner as well as vagal blockade. Neuromuscular blocking drugs that cause tachycardia are thought to have a similar mechanism of action. ACh acting on presynaptic adrenergic muscarinic receptors is a potent inhibitor of NE release.11 The prejunctional muscarinic receptor may play an important physiologic role because several autonomically innervated tissues (e.g., the heart) possess ANS plexuses in which the SNS and PNS nerve terminals are closely associated. In these plexuses, ACh, released from the nearby PNS nerve terminals (vagus nerve), can inhibit NE release by activation of presynaptic adrenergic muscarinic receptors (Fig. 15-9).
Nicotinic receptors are found at the synaptic junctions of both SNS and PNS ganglia. Because both junctions are cholinergic, ACh or ACh-like substances such as nicotine will excite
postganglionic fibers of both systems (see Fig. 15-2). Low doses of nicotine produce stimulation of ANS ganglia whereas high doses produce blockade. This dualism is referred to as the nicotinic effect (see below Ganglionic Drugs). Nicotinic stimulation of the SNS ganglia produces hypertension and tachycardia by causing the release of EPI and NE from the adrenal medulla. Adrenal hormone release is mediated by ACh in the chromaffin cells, which are analogous to postganglionic neurons (see Fig. 15-2). A further increase in nicotine concentration produces hypotension and neuromuscular weakness, as it becomes a ganglionic blocker. The cholinergic neuroeffector junction of skeletal muscle also contains nicotinic receptors, although they are not identical to the nicotinic receptors in ANS ganglia.
postganglionic fibers of both systems (see Fig. 15-2). Low doses of nicotine produce stimulation of ANS ganglia whereas high doses produce blockade. This dualism is referred to as the nicotinic effect (see below Ganglionic Drugs). Nicotinic stimulation of the SNS ganglia produces hypertension and tachycardia by causing the release of EPI and NE from the adrenal medulla. Adrenal hormone release is mediated by ACh in the chromaffin cells, which are analogous to postganglionic neurons (see Fig. 15-2). A further increase in nicotine concentration produces hypotension and neuromuscular weakness, as it becomes a ganglionic blocker. The cholinergic neuroeffector junction of skeletal muscle also contains nicotinic receptors, although they are not identical to the nicotinic receptors in ANS ganglia.
Adrenergic Receptors
Another major peripheral adrenergic receptor specific for dopamine is termed the dopaminergic (DA) receptor. Further studies have revealed not only subsets of the α and β receptors but also the DA receptor. These DA receptors have been identified in the CNS and in renal, mesenteric, and coronary vessels. The physiologic importance of these receptors is a matter of controversy because there are no identifiable peripheral DA neurons. Dopamine measured in the circulation is assumed to result from spillover from the brain.
The function of dopamine in the CNS has long been known, but the peripheral dopamine receptor has been elucidated only within the past 25 years. The presence of the peripheral DA receptor was obscured because dopamine does not affect the DA receptor exclusively. It also stimulates α and β receptors in a dose-related manner. However, DA receptors function independently of α or β blockade and are modified by DA antagonists such as haloperidol, droperidol, and phenothiazines. Thus, there is a necessity for the addition of the DA receptor and its subsets (DA1 and DA2).
The distribution of adrenergic receptors in organs and tissues is not uniform and their function differs not only by their location but also in their numbers and/or distribution. Adrenergic receptors are found in two loci in the sympathetic neuroeffector junction. They are found in both the presynaptic (prejunctional) and postsynaptic (postjunctional) sites as well as extrasynaptic sites (Fig. 15-10). Table 15-3 is a review of the function and synaptic location of some of the clinically important receptors and their subtypes.
Alpha-adrenergic Receptors
The alpha-adrenergic (α) receptors have been further subdivided into two clinically important classes, α1 and α2. This classification is based on their response to the α-antagonists yohimbine and prazosin. Prazosin is a more potent antagonist of α1 receptors, whereas α2 receptors are more sensitive to yohimbine. Recently, pharmacologic experiments have demonstrated the existence of two subtypes within the α1 group, namely α1A and α1B, and at least two subtypes within the α2, group respectively α2A, and α2B. The importance of these subsets is still emerging, with evidence that the spleen and liver contain mainly α1B receptors, and the heart, neocortex, kidney, vas deferens, and hippocampus contain equal amounts of α1A and α1B receptors. The α1-adrenergic receptors are found in the smooth muscle cells of the peripheral vasculature coronary arteries, skin, uterus, intestinal mucosa, and splanchnic beds12 (see Table 15-4). The α1 receptors serve as postsynaptic activators of vascular and intestinal smooth muscle as well as of endocrine glands. Their activation results in either decreased or increased tone, depending upon the effector organ. The response in resistance and capacitance vessels is constriction, whereas in the intestinal tract it is relaxation. There is now a large body of evidence documenting the presence of postjunctional α1 adrenoreceptors in the mammalian heart. α1-adrenergic receptors have been shown to have a positive inotropic effect on cardiac tissues in most mammals studied, including humans. Experimental work strongly supports the concept that enhanced myocardial α1 responsiveness plays a primary role in the genesis of malignant arrhythmias induced by catecholamines during myocardial ischemia and reperfusion. Drugs possessing potent α1-antagonist activity such as prazosin and phentolamine provide significant antiarrhythmic activity. The clinical mechanism and significance of these findings are not yet clear. However, there is no doubt that α1-adrenergic antagonists prevent catecholamine-induced ventricular arrhythmias.13 In contrast, studies of the effects of β-antagonists in experimental and clinical myocardial infarction have provided conflicting results.
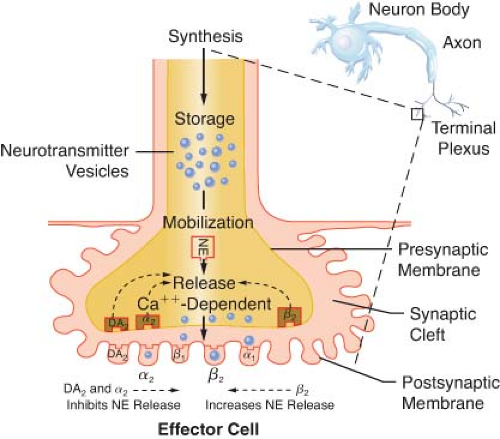 Figure 15.10. Location of several known adrenergic receptors. The presynaptic α-2 and DA receptors serve as a negative feedback mechanism, whereby stimulation of NE inhibits its own release. Presynaptic β-2 stimulation increases NE uptake, augmenting its availability. Postsynaptic α-2 and β-2 receptors are extrasynaptic and are considered non-innervated hormonal receptors. DA, dopamine; NE, norepinephrine. |
The discovery of presynaptic α-adrenoreceptors and their role in the modulation of NE transmission provided the stimulus for the subclassification of α receptors into α1 and α2 subtypes. Presynaptic α1 receptors have not been identified receptors appear confined only to the postsynaptic membrane. On the other hand, α2 receptors are found on both presynaptic and postsynaptic
membranes of the adrenergic neuroeffector junction. Table 15-4 reviews these sites. Postsynaptic membranes contain a near equal mix of α1 and α2 receptors.
membranes of the adrenergic neuroeffector junction. Table 15-4 reviews these sites. Postsynaptic membranes contain a near equal mix of α1 and α2 receptors.
Table 15-3. Adrenergic Receptors: Order of Potency of Agonists and Antagonists | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 15-4. Adrenergic Receptors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The α2 adrenoreceptors may be subdivided even further into as many as four possible subtypes. The postsynaptic α2 receptors have many actions, which include arterial and venous vasoconstriction, platelet aggregation, inhibition of insulin release, inhibition of bowel motility, stimulation of growth hormone release, and inhibition of antidiuretic hormone (ADH) release.
α2 receptors can be found in cholinergic pathways as well as in adrenergic pathways. They can significantly modulate parasympathetic activity as well. Current research implies that α2 stimulation of the parasympathetic pathways plays a role in the modulation of the baroreceptor reflex (increased sensitivity), vagal mediation of heart rate (bradycardia), bronchoconstriction, and salivation (dry mouth). However, cholinergic receptors can also be found in adrenergic pathways; thus, muscarinic and nicotinic receptors have been found in presynaptic and postsynaptic
locations, where in turn they modulate sympathetic activity (see Fig. 15-9). There is speculation that the features that are so desirable to the anesthesiologist, such as sedation, anxiolysis, analgesia, and hypnosis, are mediated through this site.
locations, where in turn they modulate sympathetic activity (see Fig. 15-9). There is speculation that the features that are so desirable to the anesthesiologist, such as sedation, anxiolysis, analgesia, and hypnosis, are mediated through this site.
Stimulation of presynaptic α2 receptors mediates inhibition of NE release into the synaptic cleft, serving as a negative feedback mechanism. The central effects are primarily related to a reduction in sympathetic outflow with a concomitantly enhanced parasympathetic outflow (e.g., enhanced baroreceptor activity). This results in a decreased systemic vascular resistance, decreased cardiac output (CO), decreased inotropic state in the myocardium, and decreased HR. The peripheral presynaptic α2 effects are similar, and NE release is inhibited in postganglionic neurons. However, stimulation of postsynaptic α2 receptors, like the α1 postsynaptic receptor, affects vasoconstriction. NE acts on both α1 and α2 receptors. Thus, NE not only activates smooth muscle vasoconstriction (postsynaptic α1 and α2 receptors) but also stimulates presynaptic α2 receptors and inhibits its own release. Selective stimulation of the presynaptic α2 receptor could produce a beneficial reduction of peripheral vascular resistance. Unfortunately, most known presynaptic α2 agonists also stimulate the postsynaptic α2 receptors, causing vasoconstriction. Blockade of α2 presynaptic receptors, however, ablates normal inhibition of NE, causing vasoconstriction. Vasodilation occurs with the blockade of postsynaptic α1 and α2 receptors.
Alpha-adrenergic Receptors in the Cardiovascular System
Postsynaptic α1 and α2 receptors in the mammalian myocardium and coronary arteries mediate a number of responses.
Coronary Arteries
The presence of postsynaptic α1 and α2 receptors in mammalian models has been demonstrated. Sympathetic nerves cause coronary vasoconstriction, which is mediated predominately by postsynaptic α2, more so than α1 receptors. The larger epicardial arteries possess mainly α1 receptors, whereas α2 receptors and some α1 receptors are present in the small coronary artery resistance vessels.14 Epicardial vessels contribute only 5% to the total resistance of the coronary circulation; therefore, α1 agonists such as phenylephrine have little influence on coronary resistance.15,16 Myocardial ischemia has been shown to increase α2 receptor density in the coronary arteries. Ischemia has also been shown to cause a reflex increase in sympathetic activity mediated by α mechanisms. This cascade may further increase coronary constriction. Postsynaptic α1 receptors do not rely upon extracellular Ca2+ to constrict the vessel, whereas the α2-constrictor response is highly dependent upon extracellular influx and exquisitely sensitive to calcium channel inhibitors.17
Myocardium
The role of β receptors in mediating catecholamine-induced inotropism and arrhythmogenesis is well known (see below Beta-adrenergic Receptors). Studies have shown the presence of postsynaptic myocardial α1 receptors, which also exert a major, facilatory, positive inotropic effect on the myocardium of several species of mammals including humans. Their contribution to malignant reperfusion arrhythmogenesis has also been recognized.
Phenylephrine, an α1 agonist, can increase myocardial contractility two- to threefold compared with a six- to sevenfold increase produced by isoproterenol, a pure β agonist. Myocardial postsynaptic α1 receptors mediate perhaps as much as 30% to 50% of the basal inotropic tone of the normal heart.
Postsynaptic myocardial α1 receptors play a more prominent inotropic role in the failing heart by serving as a reserve to the normally predominant β1 receptors. Although the response to both α1 and β1 agonists is reduced in the failing myocardium, the interaction between the two receptors is more apparent. Chronic heart failure is known to produce a reduced density (downregulation) of myocardial β1 receptors as a result of high levels of circulating catecholamines. However, there is no evidence of downregulation of either α1 or β2 receptors in cardiac failure. The increase in density of myocardial α1 adrenoreceptors is more pronounced with failure and myocardial ischemia.18 Thus, enhanced myocardial α1-receptor numbers and sensitivity may contribute to the positive inotropism seen during ischemia as well as to the malignant arrhythmias that occur with reperfusion. Intracellular mobilization of cytosolic Ca2+ by the activated α1-myocardial receptors during ischemia appears to contribute to these arrhythmias. The α1 receptor also increases the sensitivity of the contractile elements to Ca2+. Drugs possessing potent α1 antagonism such as prazosin and phentolamine have been shown to possess significant antiarrhythmic activity, but are of limited usefulness because of hypotension. Enhanced α1 activity with myocardial ischemia may explain why the antiarrhythmic benefits of β antagonists in patients with acute myocardial infarction are far from certain. The contribution of β receptors to positive inotropism and arrhythmogenesis during ischemia and reperfusion may be overshadowed by the α receptors during acute failure and ischemia.
Peripheral Vessels
Activation of the presynaptic α2-vascular receptors produces vasodilation, whereas the postsynaptic α1– and α2-vascular receptors subserve vasoconstriction. Presynaptic vascular α2 receptors inhibit NE release. This represents a negative feedback mechanism by which NE inhibits its own release via the prejunctional receptor. Presynaptic α2 agonists, such as clonidine, inhibit NE release at the neurosympathetic junction producing vasodilatation. The effect of selective presynaptic α2-receptor agonists to ameliorate coronary vasoconstriction in humans is unclear. Excitation of the inhibitory presynaptic α2 receptors by endogenous or synthetic catecholamines also inhibits NE release. However, most sympathomimetics are nonselective α agonists that will excite equally presynaptic α2 vasodilating receptors and vasoconstrictive postsynaptic α1 and α2 receptors. Postsynaptic α1 and α2 receptors coexist in both the arterial and venous sides of the circulation with the relative distribution of α2 receptors being greater on the venous side.12 This may explain why pure α1 agonists, such as methoxamine, produce little venoconstriction, whereas many nonselective agonists such as phenylephrine produce significant venoconstriction. NE is the most potent venoconstrictor of all the catecholamines. Clinically, venoconstriction would have the effect of preloading by shifting venous capacitance centrally, whereas stimulation of arterial postsynaptic α1 and α2 receptors would affect afterloading by increasing arterial resistance.
Alpha-adrenergic Receptors in the Central Nervous System
All subtypes of the α, β, and DA receptors have been found in various regions of the brain and spinal cord. The functional role of the cerebral α and β receptors suggests a close association with blood pressure and HR control. Cerebral and spinal cord presynaptic α2 receptors are also involved in inhibition of presynaptic NE release. Although the brain contains adrenergic and dopaminergic receptors, circulating catecholamines do not cross the blood–brain barrier. The catecholamines in the brain are synthesized there. Many actions have been attributed to the cerebral postsynaptic α2 receptor. This includes inhibition of insulin release, inhibition of bowel motility, stimulation of growth
hormone release, and inhibition of ADH release. Central neuraxial injections of α2 agonists, such as clonidine, induce analgesia, sedation, and cardiovascular depression. The increased duration of epidural or intrathecal anesthesia by the addition of nonselective α agonists to the local anesthetic may produce additional analgesia through this mechanism.
hormone release, and inhibition of ADH release. Central neuraxial injections of α2 agonists, such as clonidine, induce analgesia, sedation, and cardiovascular depression. The increased duration of epidural or intrathecal anesthesia by the addition of nonselective α agonists to the local anesthetic may produce additional analgesia through this mechanism.
Alpha Receptors in the Kidney
The kidney has an extensive and exclusive adrenergic innervation of the afferent and efferent glomerular arterioles, proximal and distal renal tubules, ascending loop of Henle, and juxtaglomerular apparatus. The greatest density of innervation is in the thick ascending loop of Henle, followed by the distal convoluted tubules and proximal tube. Both α1 and α2 subtypes are found in the kidney with the α2 receptor dominating. The α1 receptor is predominant in the renal vasculature and elicits vasoconstriction, which modulates renal blood flow. Tubular α1 receptors enhance sodium and water reabsorption, leading to antinatriuresis, whereas tubular α2 receptors promote sodium and water excretion.
Beta-adrenergic Receptors
The β-adrenergic receptors, like the α receptor, have been divided into subtypes. They are designated as the β1 and β2 subtypes. Recently, molecular cloning has demonstrated the existence of a third subtype, namely β3 receptor. Activation of all these receptors subtypes induces the activation of adenylyl cyclase and increased conversion of ATP to cyclic adenosine-3′,5′-monophosphate (cAMP). β1 receptors predominate in the myocardium, the sinoatrial node, and the ventricular conduction system. The β1 receptors also mediate the effects of the catecholamines on the myocardium. These receptors are equally sensitive to EPI and NE, which distinguishes them from the β2 receptors. Effects of β1 stimulation are outlined in Table 15-4, which include their effects specifically on the cardiovascular system.
The β2 receptors are located in the smooth muscles of the blood vessels in the skin, muscle, mesentery, and in bronchial smooth muscle. Stimulation produces vasodilation and bronchial relaxation. The β2 receptors are more sensitive to EPI than NE. β receptors are found in both presynaptic and postsynaptic membranes of the adrenergic neuroeffector junction (Table 15-4). β1 receptors are distributed to postsynaptic sites and have not been identified on the presynaptic membrane. Presynaptic β receptors are of the β2 subtype. The effects of activation of the presynaptic β2 receptor are diametrically opposed to those of the presynaptic α2 receptor. The presynaptic β2 receptor accelerates endogenous NE release, whereas blockade of this receptor will inhibit NE release. Antagonism of the presynaptic β2 receptors produces a physiologic result similar to activation of the presynaptic α2 receptor. The postsynaptic β1 receptors are located on the synaptic membrane and respond primarily to neuronal NE. The postsynaptic β2 receptors, like the postsynaptic α2 receptor, respond primarily to circulating EPI.
Beta Receptors in the Cardiovascular System
Myocardium
Myocardial β receptors were originally classified as β1 receptors. Those in the vascular and bronchial smooth muscle were called the β2 subtype. However, studies have confirmed the coexistence of β1 and β2 receptors in the myocardium.19 Both β1 and β2 receptors are functionally coupled to adenylate cyclase, suggesting a similar involvement in the regulation of inotropism and chronotropism. Postsynaptic β1 receptors are distributed predominantly to the myocardium, the sinoatrial node, and the ventricular conduction system. The β2 receptors have the same distribution but are presynaptic. Activation of the presynaptic β2 receptor accelerates the release of NE into the synaptic cleft. The β2 receptor comprises 20% to 30% of the β receptors in the ventricular myocardium and up to 40% of the β receptors in the atrium.
The effect of NE on inotropism in the normal heart is mediated entirely through the postsynaptic β1 receptor, whereas the inotropic effects of EPI are mediated through both the β1– and β2-myocardial receptors. The β2 receptors may also mediate the chronotropic responses to EPI which explains why selective β1 antagonists are less effective in suppressing induced tachycardia than the nonselective β1 antagonist propranolol.
Peripheral Vessels
The postsynaptic vascular β receptors are virtually all of the β2 subtype. The β2 receptors are located in the smooth muscle of the blood vessels of the skin, muscle, mesentery, and bronchi. Stimulation of the postsynaptic β2 receptor produces vasodilation and bronchial relaxation. Modest vasoconstriction occurs when subjected to blockade because the actions of the vascular postsynaptic β2 receptors no longer oppose the actions of the α1– and α2-postsynaptic receptors.
Beta Receptors in the Kidney
The kidney contains both β1 and β2 receptors with the β1 being predominant. Renin release from the juxtaglomerular apparatus is enhanced by β stimulation. The β1 receptor evokes renin release in humans. Renal β2 receptors also appear to regulate renal blood flow at the vascular level. They have been identified pharmacologically and mediate a vasodilatory response.
Dopaminergic Receptors
Dopamine, synthesized in 1910, was recognized in 1959 not only as a vasopressor and the precursor of NE and EPI, but also as an important central and peripheral neurotransmitter. Dopamine receptors (DA) are localized in the CNS, on blood vessels, and postganglionic sympathetic nerves (Table 15-4). Two clinically important types of DA receptors have been recognized: DA1 and DA2, while other subtypes such as DA4 and DA5 are still being investigated. The DA1 receptors are postsynaptic, whereas the DA2 receptors are both presynaptic and postsynaptic. The presynaptic DA2 receptors, like the presynaptic α2 receptor, inhibit NE release and can produce vasodilatation. The postsynaptic DA2 receptor may subserve vasoconstriction similar to that of the postsynaptic α2 receptor. This effect is opposite to that of the postsynaptic DA1 renal vascular receptor. The zona glomerulosa of the adrenal cortex also contains DA2 receptors, which inhibit the release of aldosterone.
Myocardium
Defining specific dopaminergic receptors has been difficult because dopamine also exerts effects on the α and β receptors. DA receptors have not been described in the myocardium. Effects of dopamine are those related to activation of β1 receptors, which promote positive inotropism and chronotropism. β2 activation may produce some systemic vasodilatation.
Peripheral Vessels
The greatest numbers of DA1-postsynaptic receptors are found on vascular smooth muscle cells of the kidney and mesentery, but are also found in the other systemic arteries including coronary, cerebral, and cutaneous arteries. The vascular receptors are, like the β2 receptors, linked to adenylate cyclase and mediate smooth muscle relaxation. Activation of these receptors produces vasodilatation, increasing blood flow to these organs. Concurrent activation of vascular presynaptic DA2 receptors also inhibits NE release at presynaptic α2 receptors, which may also contribute to peripheral vasodilatation. Higher doses of dopamine can mediate vasoconstriction via the postsynaptic α1 and α2 receptors. The constrictive effect is relatively weak in the cardiovascular system where the action of dopamine on adrenergic receptors is 1/35 and 1/50 as potent as that of EPI and NE, respectively.20
Central Nervous System
DA receptors have been identified in the hypothalamus where they are involved in prolactin release. They are also found in the basal ganglia where they coordinate motor function. Degeneration of dopaminergic neurons in the substantia nigra is the cause of Parkinson’s disease. Another central action of dopamine is to stimulate the chemoreceptor trigger zone of the medulla, producing nausea and vomiting. Dopamine antagonists such as haloperidol and droperidol are clinically effective in countering this action.
Kidney and Mesentery
Apart from their effect on the vessels of the kidney and mesentery, DA receptors on the smooth muscle of the esophagus, stomach, and small intestine enhance secretion production and reduce intestinal motility.20,21 Metoclopramide, a dopamine antagonist, is useful for aspiration prophylaxis by promoting gastric emptying. The distribution of DA receptors in the renal vasculature is well known, but DA receptors have other functions within the kidney. DA1 receptors are located on renal tubules, which inhibit sodium reabsorption with subsequent natriuresis and diuresis. The natriuresis may be the result of a combined renal vasodilatation, improved CO, and tubular action of the DA1 receptors. Juxtaglomerular cells also contain DA1 receptors, which increase renin release when activated. This action modulates the diuresis produced by DA1 activation of the tubules.
Dopamine has unique autonomic effects by activating specific peripheral dopaminergic receptors, which promote natriuresis and reduce afterload via dilatation of the renal and mesenteric arterial beds. Peripheral dopaminergic activity serves as a natural antihypertensive mechanism. Its actions are overshadowed by the opposite effect of its main biologic partner, NE. Plasma NE levels are known to increase with aging, likely the result of reduced clearance, while peripheral dopaminergic activity is known to diminish. Subtle changes in the DA–NE balance with aging may account for the diminished ability of the aged kidney to excrete a salt load.
Other Receptors
Adenosine Receptors
Adenosine produces inhibition of NE release. The effect of adenosine is blocked by caffeine and other methylxanthines. The physiologic function of these receptors may be the reduction of sympathetic tone under hypoxic conditions when adenosine production is enhanced. As a consequence of reduced NE release, cardiac work would be decreased and oxygen demand reduced. Adenosine has been effectively used to produce controlled hypotension.22
Serotonin
Serotonin (5-hydroxytryptamine) depresses the response of isolated blood vessels to SNS stimulation and decreases release of labeled NE in these preparations. Raising the external calcium ion concentration antagonizes this inhibitory action of serotonin. Thus, serotonin may inhibit neuronal NE release by a mechanism that limits the availability of calcium ions at the nerve terminal.
Prostaglandin E2, Histamine, and Opioids
Prostaglandin E2, histamine, and several opioids have been reported to act on prejunctional receptor sites to inhibit NE release in certain sympathetically innervated tissue. However, these inhibitory receptors are unlikely to play a physiologic role in limiting NE release since their direct antagonists, compounds such as inhibitors of cyclooxygenase, histamine antagonists, and naloxone do not increase a NE release.
Histamine acts in a manner similar to the neurotransmitters of the SNS. The cell membrane has specific receptors for histamine, with the individual response being determined by the type of cell being stimulated (see Chapter 12 The Allergic Response). Two receptors for histamine have been determined. These have been designated H1 and H2, for which it has been possible to develop specific agonists and antagonists. Stimulation of the H1 receptors produces bronchoconstriction and intestinal contraction. The major role of the H2 receptors is related to acid production by the parietal cells of the stomach; however, histamine is also present in relatively high concentrations in the myocardium and cardiac conducting tissue, where it exerts positive inotropic and chronotropic effects while depressing dromotropism. The positive inotropic and chronotropic effects of histamine are H2 receptor effects that are not blocked by β antagonism. These effects are blocked by H2 antagonists, such as cimetidine, which accounts for the occasional report of cardiovascular collapse following the use of cimetidine. The negative dromotropic effect and that of coronary spasm caused by histamine are H1 receptor effects.
Adrenergic Receptor Numbers and Sensitivity
Receptors, once thought to be static entities, are now thought to be dynamically regulated by a variety of conditions and to be in a constant state of flux. Receptors are synthesized in the sarcoplasmic reticulum (SR) of the parent cell, where they may remain extrasynaptic or externalize to the synaptic membranes where they may cluster. Membrane receptors may be removed or internalized to intracellular sites for either dehydration or recycling.
Principles of Clinical Pharmacology). This is thought to explain the diminished inotropic and chronotropic response to β1 agonists and exercise in patients with chronic heart failure. However, calcium-induced inotropism is not impaired because extrasynaptic β2-receptor numbers remain relatively intact. The β2 receptors may account for up to 40% of the inotropism of the failing heart compared with 20% in the normal heart.18,23 Tachyphylaxis to infused catecholamines is also thought to be the result of acute “downregulation” of receptors. There appears to be a reduction in numbers or sensitivity of β receptors in hypertensive patients who also have elevated plasma catecholamines. Downregulation is the presumptive explanation for the lack of correlation between plasma catecholamine levels and the blood pressure elevation in patients with pheochromocytoma. Chronic use of β agonists such as terbutaline, isoproterenol, or EPI for the treatment of asthma can result in tachyphylaxis because of downregulation. Even short-term use (1 to 6 hours) of β agonists may cause downregulation of receptor numbers. Downregulation is reversible on termination of the agonist. Chronic treatment of animals with nonselective β blockade causes a 100% increase in the number of β receptors. This accounts for the propranolol withdrawal syndrome in which the acute discontinuation of the β antagonist leaves the α receptors unopposed, in addition to an increased number of β receptors. Clonidine withdrawal can be explained by the same mechanism. Up- or downregulation of receptor numbers may not alter sensitivity of the receptor. Likewise, sensitivity may be increased or decreased in the presence of normal numbers of receptors. The pharmacologic factors affecting up- or downregulation of the α and β receptors are similar.
Autonomic Nervous System Reflexes and Interactions
Baroreceptors
Several reflexes in the cardiovascular system help govern arterial blood pressure control cardiac output (CO), and heart rate (HR). The aim of the circulation is to provide blood flow to all the body organs (see Chapter 10 Cardiac Anatomy and Physiology). Yet, the most important controlled variable to which the sensors are attuned is blood pressure, a product of the blood flow and vascular resistance. Étienne Marey noted in 1859 that the pulse rate is inversely proportional to the blood pressure, and this is known as Marey’s law. Subsequently, Hering, Koch, and others demonstrated that the alterations in HR evoked by changes in blood pressure are dependent on baroreceptors located in the aortic arch and the carotid sinuses. These pressure sensors react to alterations in stretch caused by blood pressure. Impulses from the carotid sinus and aortic arch reach the medullary vasomotor center by the glossopharyngeal and vagus nerves, respectively. Increased sensory traffic from the baroreceptors, caused by increased blood pressure, inhibits SNS effector traffic. The relative increase in vagal tone produces vasodilation, slowing of the HR, and a lowering of blood pressure. Real increases in vagal tone occur when blood pressure exceeds normal limits. The Valsalva maneuver can best demonstrate the arterial baroreceptor reflex (Fig. 15-11). The Valsalva maneuver raises the intrathoracic pressure by forced expiration against a closed glottis. The arterial blood pressure rises momentarily as the intrathoracic blood is forced into the heart (increased preload). Sustained intrathoracic pressure diminishes venous return, reduces the CO, and drops the blood pressure. Reflex vasoconstriction and tachycardia ensue. Blood pressure returns to normal with release of the forced expiration, but then briefly “overshoots” because of the vasoconstriction and increased venous return. A slowing of the HR accompanies the overshoot in pressure. The cardiovascular responses to the Valsalva maneuver require an intact ANS circuit from peripheral sensor to peripheral adrenergic receptors. The Valsalva maneuver has been used to identify patients at risk for anesthesia due to ANS instability (Fig. 15-11). This was once a major concern in patients receiving drugs that depleted catecholamines, such as reserpine. Dysfunction of the SNS is implicated if exaggerated and prolonged hypotension develops during the forced expiration phase (50% from resting mean arterial pressure). In addition, the overshoot at the end of the Valsalva maneuver is absent. Dysfunction of the PNS can be assumed if the HR does not respond appropriately to the blood pressure changes.
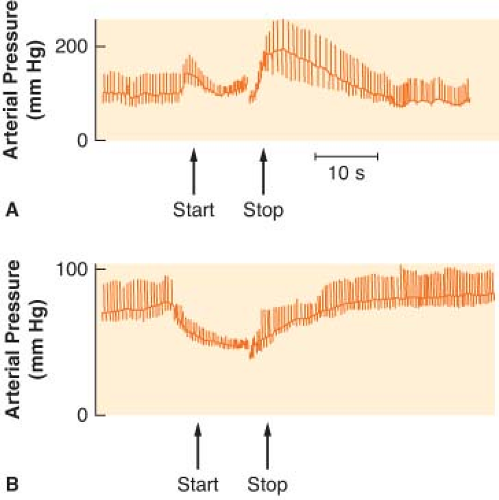 Figure 15.11. A: The normal blood pressure response to the Valsalva maneuver is demonstrated. Pulse rate moves in a reciprocal direction according to Marey’s law of the heart. B: An abnormal Valsalva response is shown in a patient with C5 quadriplegia. |
Venous baroreceptors may be more dominant in the moment-to-moment regulation of CO. Baroreceptors in the right atrium and great veins produce an increase in HR when stretched by increased right atrial pressure. Reduced venous pressure decreases HR. Unlike the arterial baroreceptors, venous sensors are not thought to alter vascular tone; however, venoconstriction is postulated to occur when atrial pressures decline. Stretch of the venous receptors produces changes in HR opposite to those produced when the arterial pressure sensors are stimulated. The
arterial and venous pressure receptors are separately monitoring two of the four major determinants of CO: Afterload and preload, respectively. Venous baroreceptors sample preload by stretch of the atrium. Arterial baroreceptors survey resistance, or afterload, as reflected in the mean arterial pressure. Afterload and preload produce opposite effects on CO; thus, one should not be surprised that the venous and arterial baroreceptors produce opposing effects after a similar stretch stimulus, pressure.
arterial and venous pressure receptors are separately monitoring two of the four major determinants of CO: Afterload and preload, respectively. Venous baroreceptors sample preload by stretch of the atrium. Arterial baroreceptors survey resistance, or afterload, as reflected in the mean arterial pressure. Afterload and preload produce opposite effects on CO; thus, one should not be surprised that the venous and arterial baroreceptors produce opposing effects after a similar stretch stimulus, pressure.
Bainbridge described the venous baroreceptor reflex and demonstrated that it can be abolished by vagal resection. Numerous investigators have confirmed the acceleration of the HR in response to volume. However, the magnitude and direction of the HR response are dependent on the prevailing HR at the time of stimulation. The denervated, transplanted mammalian heart also accelerates in response to volume loading. HR, like CO, can apparently be adjusted to the quantity of blood entering the heart. The Bainbridge reflex relates to the characteristic but paradoxical slowing of the heart seen with spinal anesthesia. Blockade of the SNS levels of T1–4 ablates the efferent limb of the cardiac accelerator nerves. This source of cardiac deceleration is obvious, as the vagus nerve is unopposed. However, bradycardia during spinal anesthesia is more related to the development of arterial hypotension than to the height of the block. The primary defect in the development of spinal hypotension is a decrease in venous return. Theoretically, the arterial hypotension should reflexly produce a tachycardia through the arterial baroreceptors. Instead, bradycardia is more common. Greene suggests that in the unmedicated person, the venous baroreceptors are dominant over the arterial. A reduced venous pressure, therefore, slows HR.24 In contrast, humorally mediated tachycardia is the usual response to hypotension or acidosis from other causes. In patients with difficult to control blood pressure, decreasing the sympathetic outflow seems to be beneficial in better regulating the blood pressure. Therefore, surgical interuption of renal efferent sympathetic outflow with radiofrequency ablation through femoral artery catheterization increases natriuresis and diuresis, and reduces renin production. Also, baroreflex sensitization through an implantable carotid sinus stimulator seems to be extremely promising in patients with refractory hypertension, with more research underway.25
Denervated Heart
Reflex modulation of the adrenergic agonists is best seen in the denervated transplant heart, which retains the recipient’s innervated sinoatrial node and the donor’s denervated sinoatrial node26 (see Chapter 51 Transplant Anesthesia). NE infusion in the transplanted heart produces a slowing of the recipient’s atrial rate through vagal feedback as the blood pressure rises. In the unmodulated donor heart, atrial rate increases. The baroreceptors are therefore not operant in the transplanted heart. Isoproterenol, a pure β agonist, increases the discharge rate of both the recipient and donor node by direct action, with the donor rate near doubling that of the recipient node. Atropine accelerates the recipient’s atrial rate, whereas no effect is seen on the donor rate, which now controls HR.
β blockade produces comparable slowing of the sinoatrial node of both recipient and donor. The exercise capability of the denervated heart is conspicuously reduced by β blockade, presumably because of its reliance on circulating catecholamines. Propranolol has also been demonstrated to reduce the β response to chronotropic effects of NE and isoproterenol in the transplanted heart. The CO of the transplanted heart varies appropriately with changes in preload and afterload.
Interaction of Autonomic Nervous System Receptors
Strong interactions have been noted between SNS and PNS nerves in organs that receive dual, antagonistic innervation. Release of NE at the presynaptic terminal is modified by the PNS. For example, vagal inhibition of left ventricular contractility is accentuated as the level of SNS activity is raised. This interaction is termed accentuated antagonism and is mediated by a combination of presynaptic and postsynaptic mechanisms. The coronary arteries present an example of this phenomenon and deserve special attention.
The myocardium and coronary vessels are abundantly supplied with adrenergic and cholinergic fibers. Strong activity of both α and β receptors has been demonstrated in the coronary vascular bed. Selective stimulation of both the α1 and postsynaptic α2 receptors increases coronary vascular resistance, whereas selective α blockade eliminates this effect. Therefore, both β1 and α1 adrenoreceptors are present on coronary arteries and accessible to NE released by sympathetic nerves.6,15
The presynaptic adrenergic terminals of the myocardium and coronary vessels, like all blood vessels studied, contain muscarinic receptors.11 Recent observations confirm that muscarinic agents and vagal stimulation, acting on the presynaptic, SNS muscarinic receptor, inhibit the release of NE in a manner similar to that of the presynaptic α2 and DA2 receptors (Fig. 15-9). Conversely, blockade of the muscarinic receptors with atropine markedly augments the positive inotropic responses to catecholamines.6 Suppression of NE release explains, in part, vagal-induced attenuation of the inotropic response to strong SNS stimulation (accentuated antagonism) and only a weak negative inotropic effect of vagal stimulation when there is low background SNS activity. This may also explain why vagal activity reduces the vulnerability of the myocardium to fibrillation during infusions of NE.
ACh may cause coronary spasm during periods of high SNS tone.6 Inhibition of NE release by presynaptic adrenergic muscarinic receptors of the smooth muscle of coronary vessels would lessen the coronary relaxation normally produced by NE on the β1 receptor (Fig. 15-9). In anesthetized dogs, the rate of NE outflow into the coronary sinus blood, evoked by cardiac SNS stimulation, is markedly diminished by simultaneous vagal efferent stimulation.27 This action is known to be prevented by atropine, which also causes coronary vasodilation.
Interaction with Other Regulatory Systems
on the adrenal cortex to release aldosterone and on the adrenal medulla to release EPI. In addition to its direct effects on vascular smooth muscle, angiotensin II augments NE release via presynaptic receptors, thus enhancing peripheral SNS tone. Captopril, enalapril, and lisinopril inhibit the action of converting enzyme, thus preventing the conversion of angiotensin I to angiotensin II. Renin is released in response to hyponatremia, decreased renal perfusion pressure, and ANS stimulation via β receptors on juxtaglomerular cells. Changes in sympathetic tone may thus alter renin release and affect homeostasis in a variety of ways. The ANS is also intimately related to adrenocortical function. As outlined above, glucocorticoid release modulates phenylethanolamine-N-methyltransferase formation and thus synthesis of EPI. Glucocorticoids are also important in regulating the response of peripheral tissues to changes in SNS tone. Thus, the ANS is intimately related to other homeostatic mechanisms.
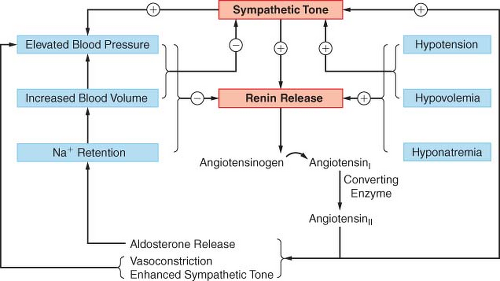 Figure 15.12. The interactions of the renin–angiotensin and SNS in regulating homeostasis are shown schematically along with the physiologic variables that modulate their function. Arrows with a plus sign (+) represent stimulation, and those with a minus sign (–) represent inhibition. |
Clinical Autonomic Nervous System Pharmacology
Mode of Action
ANS drugs may be broadly classified by mode of action according to their mimetic or lytic actions. This may also be termed agonist or antagonist. A sympathomimetic, such as ephedrine, mimics SNS sympathetic activity by stimulation of adrenergic receptor sites both directly and indirectly. Sympatholytic drugs cause dissolution of SNS activity at these same receptor sites. β receptor blockers are examples of sympatholytic drugs. Several modes of ANS drug action become evident when one follows the cascade of neurotransmission. Drugs that act on prejunctional membranes may therefore (1) interfere with transmitter synthesis (α-methyl paratyrosine), (2) interfere with transmitter storage (reserpine), (3) interfere with transmitter release (clonidine), (4) stimulate transmitter release (ephedrine), or (5) interfere with reuptake of transmitter (cocaine). Drugs may also (6) modify metabolism of the neurotransmitter in the synaptic cleft (anticholinesterase). Drugs acting at postjunctional sites may (7) directly stimulate postjunctional receptors and (8) interfere with transmitter agonist at the postjunctional receptor.
The ultimate response of an effector organ to an agonist or antagonist depends on (1) the drug, (2) its plasma concentration, (3) the number of receptors in the effector organ, (4) binding by the receptor, (5) the concurrent activities of other drugs and hormones, (6) the cellular metabolic status, and (7) reflex adjustments by the organism.
Ganglionic Drugs
SNS and PNS ganglia are pharmacologically similar in that the transmission through these ANS ganglia is effected by ACh (see Fig. 15-2). Most ganglionic agonists and antagonists are not selective and affect SNS and PNS ganglia equally. This nonselective property creates many undesirable and unpredictable side effects, which have limited the clinical usefulness of this category of drug.
Agonists
There are essentially no clinically useful ganglionic agonists. Nicotine is the prototypical ganglionic agonist. In low doses, it stimulates ANS ganglia and the neuromuscular junction of striated muscle. High doses produce ganglionic and neuromuscular blockade. The protean side effects of nicotinic stimulation render it useful only as an investigative tool.
Related posts:





Full access? Get Clinical Tree


