(1)
Northern Anesthesia & Pain Medicine, LLC, Eagle River, Alaska, USA
(2)
WWAMI Program, University of Washington School of Medicine, Anchorage, Alaska, USA
Keywords
AgentPathogenicAntibiotic resistanceVirulenceAntivirulenceOpioidAttenuationMesolimbicNucleus accumbensVentral tegmental areaExtended-releaseLong-actingControlled-releaseSustained-releaseTimed-releaseOxyContinRisk evaluation and mitigation strategy (REMS)Abuse-deterrentTamper-resistantPartial agonistBuprenorphineProbuphineEnkephalinase inhibitorProdrugBiased agonismAllosteric modifierTLR4 antagonistIbudilastAV411Two young men in a remote village were abusing an extended-release oxycodone product via crushing the pills and “smoking” the powder, inhaling the fumes via a pipe. An argument ensued between the two of them, and one of the young men shot his associate several times in the abdomen with a 9 mm pistol. By report, the two men resolved their differences shortly thereafter and resumed smoking their oxycodone together. A few hours later, the assailant turned the gun upon himself and took his own life. The man shot in the abdomen then sought medical attention and was air evacuated and transferred to the regional trauma center where he underwent exploratory laparotomy, removal of projectiles, and excision/repair of injured bowel. He survived this incident.
Introduction
One theoretical approach to end an epidemic is to eliminate the agent.
This is neither realistic nor in all likelihood would it be in the best interest of the host population, in many cases. First of all, the agents are ubiquitous. Secondly, most microbes play some role in the larger balance of the ecosystem, and their elimination may have far-reaching negative consequences. Thirdly, mutation rates coupled with short propagation times invariably result in new pathogenic strains, or antimicrobial-resistant strains, as we have learned over the last few decades in the infectious disease realm.
Many of these considerations apply to the opioid as agent as well. Blanket elimination is impossible without destroying every poppy plant and chemical laboratory on the planet. Opioids do provide a necessary and useful role, and their elimination would have far-reaching negative consequences. Human nature is such that when one abusable substance is rendered inaccessible, another is often substituted.
An alternate approach addressing the agent is to somehow attenuate its virulence without eliminating it. Since the post-World War II period and Dr. Jonas Salk’s inactivated and Dr. Albert Sabin’s “live-attenuated” poliovirus vaccines, alteration of normal microbial pathogenic capacity has been the mainstay of infectious disease prevention, by means of stimulating host immune defenses via vaccination. More recently, “antivirulence therapies ” aimed at altering agent virulence factor production, expression, regulation, etc. are under intensive research and development as classic twentieth century antimicrobial therapeutic approaches seem doomed to failure as resistance inevitably overcomes antibiotics. Some of these approaches include targeting adherence and protective (biofilm) mechanisms, enhancing competition for resources by increasing commensal organisms, interfering with “quorum sensing ” and signaling among pathogens, “quorum quenching” agents, and other novel approaches [1–3].
This sort of multifaceted “outside-the-box thinking” must be applied to the development of novel analgesics as well if we hope to reduce human dependence upon exogenous opioids. Classic approaches (such as extended-release/long-acting formulations and abuse-deterrent/tamper-resistant technologies, discussed in greater detail below) have not proven successful on a wide scale in containing the opioid epidemic.
Nora Volkow’s 2014 Congressional Address highlighted the need for the development of novel analgesic therapies that “circumvent the brain reward pathways, thereby greatly reducing abuse potential” [4]. In considering antivirulence strategies, one must first define and understand virulence. A succinct, recent definition highlighting both the concept of virulence along a spectrum and also its essentially broad context is “the relative capacity of a microbe to cause damage in a host [5].” These authors also make the point that the virulence of an agent may be dependent upon individual host vulnerability which varies as well. Host damage caused by opioids has been reviewed in Chap. 3 and ranges from mildly annoying side effects to death from respiratory depression and in some cases cardiac arrest. Significant morbidity affecting nearly every physiologic system is common but varies from individual to individual. Obese individuals with compromised airways and those with ultra-rapid CYP2D6 metabolizer status are more prone to respiratory arrest, and individuals with certain psychosocial risk factors (as discussed in greater detail in Chap. 9) are more prone to dependence and addiction. However, while the vulnerability to and outcome of opioid morbidity (including dependence and addiction) and mortality are variable, their correlation with increased exposure is not only intuitive but borne out in the literature. In addition, the correlation between abuse potential and increased exposure is well-established. Thus, while death (or addiction) may result from one single exposure and many chronic users survive and escape addiction, in general reducing abuse potential is a key strategy in attenuating virulence, as Dr. Volkow points out.
Brief Overview of Opioid Addiction Biology
Before examining past, present, and future mechanisms of reducing opioid virulence, a cursory review of current neurobiological theory of (psychological) substance dependence and addiction, along with proposed mechanisms whereby opioids “hijack” the proposed circuitry, is in order. A more comprehensive overview is presented in Chap. 9, with behavioral and societal theories broadening the discussion. It should be noted at the outset of any exploration of proposed addiction biology that:
Interspecies differences limit extrapolation of animal neuroanatomic and neurophysiologic research to humans.
There are profound and even diametrically opposed differences in response to the same stimulus within the same animal model depending on whether the subjects are awake or anesthetized [6].
Human and nonhuman primate higher function confers complexity to the process of dependence and addiction not represented in lower species. Animals are incapable of understanding negative consequences of addictive behavior, and the added layers of cognitive, emotional, relational, and spiritual stresses that addiction confers to humans are simply not represented.
Current understanding of the neurobiology of reward motivation assigns a central role to dopaminergic neurons within the ventral tegmental area (VTA) of the midbrain. VTA dopaminergic neurons project superiorly along the medial forebrain bundle (nicknamed the “hedonic highway” [7]) to various limbic and executive areas including the:
Amygdala – within the temporal lobe, this limbic structure is involved in emotional processing and memory.
Ventral striatum (nucleus accumbens, NAc) – this basal ganglia structure is held to be the central region mediating motivation for pursuing rewarding substances or activities. The pathway between VTA and NAc is known as the mesolimbic pathway (shown in green in Fig. 5.1) and has long been held to be the key link in natural and exogenous reward/reinforcement and addiction.
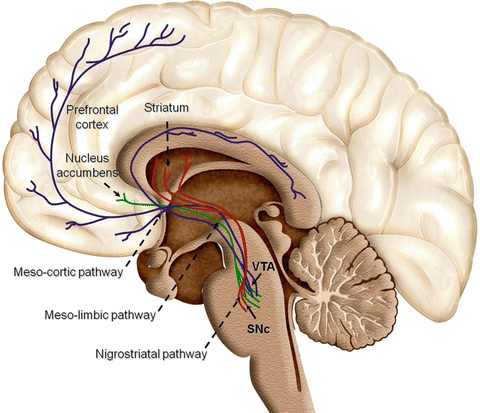
Fig. 5.1
Reward and addiction pathways in the brain. SNc, substantia nigra; VTA, ventral tegmental area. Reprinted from Ref. [8]. ©Arias-Carrión et al. (2010). http://www.biomedcentral.com/1755-7682/3/24/, CC BY 3.0, https://commons.wikimedia.org/w/index.php?curid=44695433
Anterior cingulate cortex – this paralimbic region is involved in the anticipation of reward and plays a key role in integrating emotions with painful stimuli as well.
Prefrontal cortex (PFC ) – this “executive center” of the brain is involved in decision-making based on integration of sensory and limbic inputs as well as memory and is thought to be a critical area for reinforcing drug effects.
Intracerebral dopamine agonists induce both self-administration and conditioned place preference (CPP) in rodents [9–12], and dopamine antagonism causes aversive behaviors in both animal and human subjects [13–15]. Virtually all drugs of abuse and other pleasurable stimuli are believed to exert hedonic effect via mesolimbic dopamine.
Opioid-induced reward (including euphoria) is thought to be mediated via cerebral mu-opioid receptors (MOR) [16–18]. MOR are ubiquitous throughout the brain, but extensive research in animal subjects has shown that there are specific regions, in particular the VTA, whose MOR appear particularly important to the reward/reinforcement phenomenon that is universally accepted as fundamental to the development of addiction [19–22]. Evidence for the centrality of the VTA in opioid dependence is both positive (intra-VTA opioid administration induces both self-administration and CPP in rodents [20, 22–24]) and negative (intra-VTA opioid antagonists or genetic MOR knockout results in loss of these behaviors [21, 25]).
Dopaminergic transmission to mesolimbic targets including the NAc is effected by VTA MOR agonism [26, 27], and thus a synthesis of these data has resulted in opioid-VTA MOR-mesolimbic dopaminergism being widely accepted as the foundational biologic paradigm for opioid addiction [28–30].
However, several recent lines of evidence indicate that the picture is much more complicated than this. First of all, ample evidence indicates that not all NAc dopaminergism is rewarding; several agents (delta-opioid receptor agonists, cholecystokinin, glial-derived neurotrophic factor) increase dopamine in the NAc but do not produce CPP [6], and furthermore even the administration of MOR antagonists sufficient to cause place aversion also results in NAc dopamine increases [31, 32]. Second, evidence is accumulating that opioid reinforcement can occur in the absence of mesolimbic dopaminergism [33–36]. Interestingly, dopaminergic neuronal activity in response to MOR agonism has been shown to increase in anesthetized animals but decrease in awake animals [6]. Similarly, MOR-mediated CPP inhibition by dopamine antagonism has been shown in opioid-dependent rodents whereas opioid-naïve animals do not lose CPP [37]. Finally, stimulation of VTA dopaminergic neurons has been shown to also result in release of other neurotransmitters including glutamate and GABA [38–40].
While our understanding of the complexities of addiction neurobiology (let alone behavior) is in its infancy, it is at least a useful construct to imagine a correlation between a substance’s capacity to agonize midbrain MOR and effect pleasurable dopaminergic (or other) transmission to limbic and higher cortical structures and its virulence.
Overview of Virulence Attenuation Strategies
Historic and current approaches to attenuating opioid virulence are organized in this chapter into two categories:
- 1.
Modifying existing mu-opioid receptor (MOR) agonists
- 2.
Other approaches
The second category is necessarily and by design broad, indicating the diversity of approaches available. It is also intended to highlight the distinction between “out-of-the-box” thinking and more restricted efforts typified by the first category. This is not to disparage efforts to reduce virulence by altering delivery of currently extant agents; there is great practicality in utilizing agents already known to be (relatively) safe and efficacious. Such approaches however have little promise for improvement over the current situation besides limiting the potential for non-sanctioned routes of abuse. Development of novel opioid-mediated strategies will be necessary to effectively harness the full, balanced potential of the intricate and wonderful endogenous analgesic system we possess. Development of non-opioid-mediated strategies (e.g., N-methyl d-aspartate blockers, cannabidiol analogs) of course will provide complementary or alternative means of potent analgesia but are outside the scope of this work.
Modifying Existing Central MOR Agonists
Extended-Release/Long-Acting Opioids
The development of extended-release or long-acting (ER/LA) opioids , in addition to providing more stable pharmacokinetics and theoretically improved baseline analgesia, has also been thought to reduce the virulence of opioids. Synonyms include controlled-release , sustained-release , and time-release technology . These formulations are engineered pharmaceutically to provide more stable plasma and target site concentrations of the drug over a prolonged period. Inherent in this strategy is a greater amount of active drug presented per single vehicle, which is typically intended to effect analgesia over a 12–24 h period. Common mechanisms include embedding the agent within a poorly soluble or insoluble matrix from which it must diffuse or microencapsulation of the agent with a slowly dissolving “coat.”
From a practical standpoint, individuals with malabsorptive syndromes (which are increasing in this country due to bariatric surgical operations) likely enjoy significantly less bioavailability of ER/LA formulations although there are not yet robust studies supporting this assumption.
Current ER/LA opioids in oral form include hydrocodone, hydromorphone, morphine, oxycodone, oxymorphone, tapentadol, and tramadol. Another means of achieving this sort of stable pharmacokinetics is via a transdermal delivery system, such as is used with certain lipophilic agents such as fentanyl and buprenorphine; this route circumvents the issues of oral absorption and may also confer reduced psychological dependence by eliminating pill-taking behavior.
Different arguments have been made in support of ER/LA opioid therapy, including the theory that chronic pain conditions should be treated with “round-the-clock” steady-state analgesia. From a virulence reduction standpoint, ER/LA agents result in lower peak plasma levels which have been thought to confer increased safety [41] although this may not be the case in situations of compromised metabolism, i.e., hepatic or renal insufficiency. Two large recent reviews have provided evidence that ER/LA agents may in fact be associated with a higher incidence of morbidity and mortality [42, 43].
As far as abuse liability, rapid onset or immediate-release drugs have been shown repeatedly to be preferred by abusers [44–48]—presumably as they result in more dramatic/dynamic presentation of dopamine from the VTA to the NAc and other limbic reward centers—drugs with a rapid elimination rate have also been associated with greater self-administration [44, 45]. Conversely, ER/LA formulations are believed to confer a lower risk of psychological (not physical) dependence and addiction and have been touted as conferring lower abuse liability [41, 49] although other data have called this conclusion into question as well [50, 51].
Ironically, the current opioid crisis in this country is often attributed to ER/LA oxycodone (Purdue’s OxyContin) which entered the American market in 1996. The mining injury-rich and depressed socioeconomic climate of rural Appalachia is credited with facilitating a demographic ripe for substance abuse and addiction, and here the drug enjoyed particularly rapid adoption among prescribers thanks to aggressive marketing and subsidization. Individuals quickly discovered how to defeat the time-release mechanism by crushing and insufflating or injecting the powder, delivering a large dose of oxycodone (up to 160 mg from one tablet, equivalent to over 300 mg of morphine with a typical hospital inpatient dose being 2–4 mg of intravenous morphine) instantly. Rates of abuse, overdose and death, and crime soared over the next decade in what the Office of National Drug Control Policy has termed the Appalachia High Intensity Drug Trafficking Area, which includes 67 counties in Kentucky, Tennessee, and West Virginia. OxyContin became the top-selling pharmaceutical opioid in history, earning Purdue $35 billion between 1996 and 2015 [52]. After numerous investigations and lawsuits, three top officials ultimately pleaded guilty in 2007 to misleading the public about the drug’s risk of addiction and paid $634.5 million in fines; in the late 2015 Purdue settled with the State of Kentucky for another $24 million in damages, and several other lawsuits remain in court (Fig. 5.2).
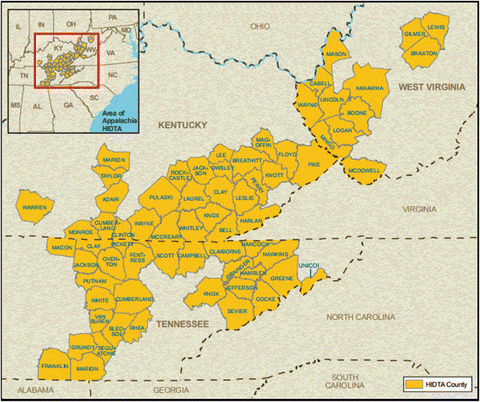
Fig. 5.2
Appalachia High Intensity Drug Trafficking Area. National Drug Intelligence Center, US Department of Justice
In 2012, the US Food and Drug Administration (FDA) initiated a Risk Evaluation and Mitigation Strategy (REMS) requiring ER/LA opioid manufacturers to make available prescribing training for healthcare professionals who prescribe ER/LA opioids and also to distribute educational materials to prescribers and patients on the safe use of these powerful pain medications.
Abuse-Deterrent/Tamper-Resistant Formulations
The OxyContin experience resulted in increased interest in/pressure to formulate higher-dose opioids in a vehicle that would prevent abusers from circumventing the time-release mechanisms, rendering them essentially immediate-release preparations of supratherapeutic doses. Common mechanisms for defeating ER/LA technology by abusers have included crushing and snorting or dissolving in water or solvents and injecting.
Abuse-deterrent or tamper-resistant strategies have multiplied over the past decade and include diverse tactics such as altering the physical formulation, adding aversive agents activated upon alteration or administration by non-indicated routes, and implant formulation in the case of buprenorphine. Currently available formulations are listed in Table 5.1.
Table 5.1
Abuse-deterrent/tamper-resistant opioids
Drug | Brand name | Mechanism of abuse deterrence/tamper resistance |
|---|---|---|
Buprenorphine | Probuphine® | Partial agonist, MOR “blocker,” subdermal implant system |
Buprenorphine-naloxone | Suboxone® | Partial agonist, MOR “blocker,” integrated naloxone |
Hydrocodone ER | Hysingla® | RESISTEC® polymer matrix controls release and renders tablet difficult to crush and forms viscous gel when dissolved in aqueous solutions, hampering injection |
Hydrocodone ER | Zohydro® | BeadTek® mixture of inactive beads, active immediate release, and active ER hydrocodone beads. Inactive beads maintain the 12 hr release properties of the drug when taken as directed but will immediately form a viscous gel when crushed and dissolved in liquids or solvents |
Hydromorphone ER | Exalgo® | OROS® osmotic delivery system facilitates controlled release and also crush and extraction resistance properties |
Morphine-naltrexone | Embeda® | Naltrexone is sequestered in micropellets’ cores and is released upon crushing |
Oxycodone | Oxaydo® | AVERSION® matrix forms viscous gelatinous mixture hampering injection; if crushed and snorted, sodium lauryl sulfate will cause nasal discomfort |
Oxycodone ER | OxyContin® | INTAC® hydrophilic matrix controls release and forms a gel that cannot be easily injected or snorted if crushed or dissolved in solutions and resists extraction of active drug via solvents |
Oxymorphone ER | Opana ER® | INTAC® hydrophilic matrix controls release and forms a gel that cannot be easily injected or snorted if crushed or dissolved in solutions and resists extraction of active drug via solvents |
Tapentadol ER | Nucynta ER® | INTAC® hydrophilic matrix controls release and forms a gel that cannot be easily injected or snorted if crushed or dissolved in solutions and resists extraction of active drug via solvents |
Tamper Resistance
Several physical component mechanisms exist to discourage tampering, including formulation as extremely hard tablets that are resistant to crushing, and embedding active drug within a matrix such as polyethylene oxide that forms a viscous gel when exposed to water or solvent, hampering injection.
According to the National Poison Data System, “abuse exposures” related to OxyContin decreased by 36% in the 1-year period following the reformulation with INTAC tamper-resistant technology [53], and according to the manufacturer’s adverse events database, related deaths are decreased by 82% from the year prior to the third year after reformulation [54].
Aversive Technology
Aversive technology has primarily taken the form of adding MOR antagonists (naloxone or naltrexone) into the formulation with the therapeutic opioid to deter administration by alternate/non-indicated routes. In the case of Suboxone® or generic buprenorphine/naloxone, sublingual (or accidental enteric) administration results in minimal antagonist activity due to the low bioavailability of naloxone by those routes; however, injection of dissolved agent results in significant antagonist effects and can precipitate profound opioid withdrawal. Similarly, in the case of Embeda® (ER/LA morphine with naltrexone), the naltrexone component is sequestered in the core of the micropellets and is released upon crushing.
The AVERSION® technology of Oxaydo® further discourages crushing and insufflation by the presence of inactive ingredients that cause nasal discomfort.
Implant Systems
While tamper-resistant technology undoubtedly discourages abuse by non-indicated routes, it does not have the capability of deterring abuse by indicated routes, i.e., individuals can ingest greater quantities orally without suffering the consequences of sequestered aversive agent release, etc. Depot/time-release intramuscular injections or implant systems are one response to this liability, although (except in the case of buprenorphine, discussed below) they still do not discourage/prevent individuals from obtaining and using other opioids.
The only currently approved opioid implant system (aside from intrathecal pump systems which are not discussed in this book) is a buprenorphine implant (Probuphine®) manufactured by Braeburn, which releases 80 mg of buprenorphine over the course of 6 months. It is a solid, matchstick-sized implant made from a mixture of ethylene-vinyl acetate (EVA) and buprenorphine. While undoubtedly advantageous in improving compliance with buprenorphine treatment for opioid dependence/reducing illicit use, this system may be problematic in terms of conferring unwanted MOR blockade in cases of trauma or emergency surgery.
Federal Oversight
The original OxyContin formulation was touted/labeled as being abuse-deterrent; after it became evident that individuals were abusing the product, Purdue removed the claim of abuse deterrence from the label, which was regranted after reformulation. Since then, the FDA has exerted significantly greater oversight into the approval and marketing of abuse-deterrent formulations and recently released guidelines on the matter to the pharmaceutical industry [55].
Required processes include:
- 1.
Laboratory-based in vitro manipulation and extraction studies (Category 1)
- 2.
Pharmacokinetic studies (Category 2)
- 3.
Clinical abuse potential studies (Category 3)
- 4.
Postmarket impact studies (Category 4)
The FDA has also stated that it will require advisory committee meetings for all new non-abuse-deterrent opioids and has made clear that their intent is to facilitate eventual transition away from non-abuse-deterrent opioids.
Prodrugs
Another approach to reducing abuse proposed by the FDA [55] is the development of prodrugs. Prodrugs are less active or inactive precursor molecules that require biophysical alteration (generally by enzymatic cleavage) for transformation into active agents [56]. Prodrugs requiring gastrointestinal enzymatic activation may reduce parenteral abuse by eliminating therapeutic or recreational efficacy by non-indicated routes. However, prodrugs yielding MOR agonists as we know them today are unlikely to reduce abuse by excessive oral administration nor inherently circumvent the adverse effect profile of excessive or unbalanced MOR activation including euphoria, respiratory depression, etc.
“Outside-the-Box” Approaches
Partial Agonists
Partial mu-opioid receptor (MOR) agonists have been explored and trialed for almost half a century as a means of reducing adverse physical or psychiatric effects. Relative agonism is defined by in vitro studies comparing agents to a “full agonist” or one with maximally known efficacy. Reduced efficacy at the MOR confers a so-called ceiling effect beyond which further administration yields insignificant or at least significantly diminishing additive benefit; adverse effects mediated by the partially activated MOR are proportionately reduced but not eliminated [57]. Enthusiasm for this class of medication in terms of reduced respiratory depression and abuse liability and dependence has waxed and waned over the years [58–62]. Butorphanol, nalbuphine, and pentazocine are three older partial MOR agonists used in various “niche” arenas (e.g., butorphanol is still commonly used as a parenteral analgesic in many labor and delivery suites; nalbuphine is still used as a parenteral antipruritic to combat neuraxial opioid-mediated pruritus by many anesthesiologists), but by and large these agents have not enjoyed widespread adoption. This is in part due to the psychomimetic effects of kappa-opioid receptor (KOR) agonism that all three agents possess.
Buprenorphine (the pharmacology of which is discussed in greater detail in Chap. 4) is a partial agonist at the MOR and an antagonist at the KOR, which latter property eliminates the psychomimetic effects associated with the agents listed above. In fact, the KOR antagonism of buprenorphine has been shown confer significant antidepressant effects, and a therapeutic indication for treatment of refractory depression has been called for by the psychiatric community [63, 64]. Buprenorphine also possesses the lowest receptor dissociation constant [65, 66] (i.e., highest tenacity for the MOR) among currently available opioids, which is also relevant in terms of reducing adverse effects in that the agent effectively “blocks” other MOR agonists from exerting effects. This has led to its widespread use in treating opioid dependence/addiction; once an individual’s MOR are occupied by buprenorphine, therapeutic or hedonic/euphoric effects of other MOR agonists including heroin are subverted.
The addition of naloxone to buprenorphine (introduced in the previous section on abuse deterrence/tamper resistance) does not alter the inherent effects conferred by its partial agonist or receptor dissociation qualities.
Peripheral Agonists
Restricting opioid agonism to the periphery is one means of limiting centrally mediated adverse effects, including euphoria and the development of dependence. Whether or not such an approach can provide meaningful analgesia remains to be seen but has been intensively investigated and remains an elegant and intriguing direction.
Peripheral opioid effects are thought to be most pronounced in states of tissue inflammation [67–69], and this phenomenon correlates with observation of increased peripheral opioid receptor synthesis/expression in states of inflammation [70–72].
Besides obviation of central activity, this “limited efficacy” may be beneficial in the sense of restricting self-administration to states of nociceptive pain where pathologic peripheral inflammation is present (e.g., rheumatoid arthritis) and reducing the desirability of the agent in states of “exclusively” centrally mediated states.
Related posts:
 Best Practices Education, Part III: Regulatory and Advisory Issues Related to Opioid Therapy for Pain
Best Practices Education, Part III: Regulatory and Advisory Issues Related to Opioid Therapy for Pain
 Best Practices Education, Part II: Evidence for and Against Opioid Therapy
Best Practices Education, Part II: Evidence for and Against Opioid Therapy
 Best Practices Education, Part I: Pain Physiology, Psychology, and Alternatives to Opioids
Best Practices Education, Part I: Pain Physiology, Psychology, and Alternatives to Opioids
 Addressing Host Factors: Primary, Secondary, and Tertiary Prevention of Opioid Dependence
Addressing Host Factors: Primary, Secondary, and Tertiary Prevention of Opioid Dependence
 Opioid Dependence Risk Factors and Risk Assessment
Opioid Dependence Risk Factors and Risk Assessment
 Addressing Host Factors: Overview of Dependence and Addiction
Addressing Host Factors: Overview of Dependence and Addiction

Full access? Get Clinical Tree


