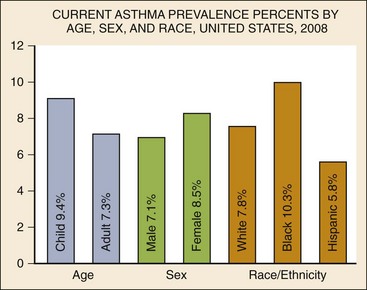
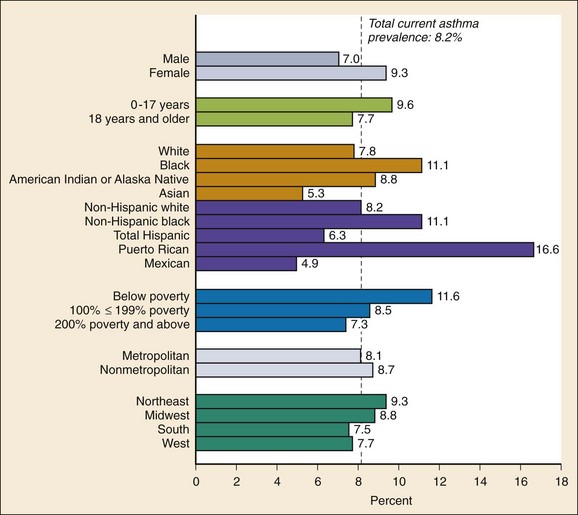
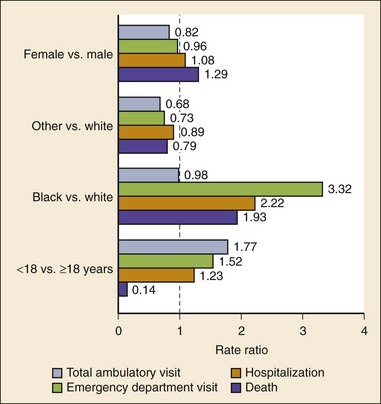
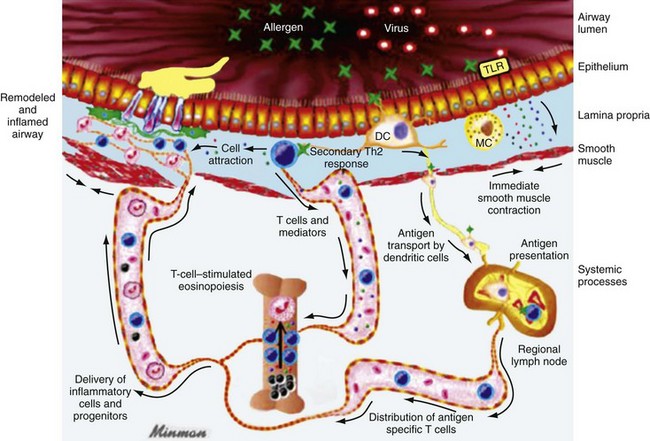
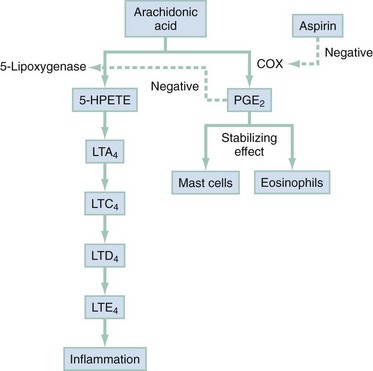
Chapter 73 The National Heart, Lung, and Blood Institute summarizes our current understanding of asthma as “a chronic inflammatory disorder of the airways in which many cells and cellular elements play a role … this inflammation causes recurrent episodes of wheezing, breathlessness, chest tightness, and coughing … episodes are usually associated with widespread but variable airflow obstruction that is often reversible either spontaneously or with treatment. The inflammation also causes an associated increase in the existing bronchial hyper-responsiveness to a variety of stimuli. Reversibility … may be incomplete in some patients.”1 Asthma is thus a chronic inflammatory disease, and control of symptoms ultimately depends on ameliorating the inflammatory reaction that produces alterations in airway function and structure. Irreversible structural airway changes occurring in response to chronic airway inflammation may influence emergency department (ED) asthma therapies. In 2008, it was estimated that 38.1 million Americans had been diagnosed with asthma by a health professional within their lifetime.2 The prevalence of asthma (defined as individuals who have been diagnosed and currently have asthma) in 2009 was 24.6 million (17.5 million adults and 7.1 million children), and the asthma attack prevalence (the number of persons who had had at least one asthma attack in the previous year) was 12.8 million, representing 52% of persons who had asthma.2,3 Asthma is more prevalent in children than adults, in females than males, and in blacks than whites or Hispanics (Fig. 73-1).4 Puerto Ricans have a current asthma prevalence 100% higher than non-Hispanic whites and 50% higher than non-Hispanic blacks. Females have a 33% higher prevalence than males (this pattern is reversed in children). Asthma is more prevalent in the impoverished and in those residing in nonmetropolitan locales. The northeastern states have the highest asthma prevalence (Fig. 73-2).3,5 Figure 73-1 Asthma prevalence percentages by age, sex, and race, United States, 2008. (Adapted from National Health Interview Survey, National Center for Health Statistics, Centers for Disease Control and Prevention. www.cdc.gov/asthma/asthmadata.htm.) Asthma was responsible for more than 1.75 million ED visits and 456,000 hospitalizations in the United States in 2007 (Fig. 73-3).3 Females were 1.08 times more likely to require hospitalization and had a death rate 1.29 times greater than males. African Americans had an ED visit rate 3.32 times higher, a hospitalization rate 2.22 times higher, and a death rate 1.93 times higher than whites. Children (ages 0-17) accounted for more than 640,000 ED visits and had a hospitalization rate 1.23 times higher than adults. Decreases in hospitalization rates have been noted in those older than 45 years from the late 1980s into the early 2000s.6 Between 1995 and 2006, hospital discharges for asthma decreased by 13%. The only age group that did not experience a decrease in hospital discharge rates was patients older than 65 years.2 The estimated financial burden of asthma totaled $20.7 billion in the United States in 2010, with approximately 75% attributable to direct costs (hospital care and physician services).2 Prescription drugs represented the highest single direct cost at $5.9 billion. Less than 20% of asthma patients account for 80% of the direct costs.7 Indirect costs attributable to asthma are reflected by decreased productivity. In 2008, asthma accounted for an estimated 14.2 million lost work days and 14.4 million lost school days. In those not currently employed, 22 million days of housework or other activities were missed.3,8 Asthma ranks within the top 10 prevalent conditions causing limitation of activity. The United States has one of the lowest rates of asthma mortality in the world.9 A downward trend in asthma-related deaths has occurred since 2003, when 4055 deaths from asthma were reported10; 3447 deaths from asthma occurred in 2007, and 3395 in 2008.11 Despite these positive indicators, asthma mortality is 44% higher in females than in males and three times higher in blacks than in whites.2 Asthma death rates are higher in Puerto Rico than in the U.S. general population.12 Deaths from asthma are rare in children.3 Evidence that inflammation is a component of asthma physiology was initially derived from autopsy findings in patients with fatal asthma. The airways revealed infiltration by neutrophils, eosinophils, and mast cells and the presence of subbasement membrane thickening, loss of epithelial cell integrity, goblet cell hyperplasia, and mucous plugs. Antemortem bronchial biopsy findings in patients with even mild degrees of asthma also demonstrate inflammatory changes in the central and peripheral airways that correlate with disease severity.13 Inflammatory and chemotactic cytokines produced by both resident airway and recruited inflammatory cells are identified in bronchoalveolar lavage washings and pulmonary secretions. Asthma has been divided into allergic and nonallergic types based on the presence or absence of immunoglobulin E (IgE) antibodies to common environmental antigens (pollen, dander, mites) and microbiologic antigens (bacteria, viruses). Immunologic responses suggest an acquired cause of asthma, whereas the absence of an immunologic response suggests an innate cause. Exposure to microbes and allergens (or lack of) during childbirth, infancy, and childhood may influence expression of the asthma phenotype later in life (the hygiene hypothesis).14–16 Trigger factors precipitating acute asthma exacerbations are typically environmental or viruses, but a specific inciting agent may not be identifiable. Regardless of an allergic or nonallergic basis, both forms of asthma are characterized by infiltration of the airways by T-helper cells that release cytokines such as interleukin (IL)-4, IL-5, and IL-13, stimulating basophil, eosinophil, mast cell, and leukocyte migration to the airways. Release of inflammatory cell mediators results in bronchospasm, mucous production, airway edema, and amplification of the inflammatory response. Mast cells are found in airway mucosa and submucosa, and increased numbers are identified in and around airway smooth muscle and airway mucous glands. Mast cell activation occurs via cross-linking of antigen and surface-bound IgE. Nonimmunologic activation by cytokines, complement, proteases, and local hyperosmolality also occurs.17 Mast cells release histamine, prostaglandin D2 (PGD2), and leukotriene C4 (LTC4), leading to bronchospasm, mucous production, and airway edema and causing acute symptoms (the early asthmatic response). Cytokines produced by mast cells result in airway smooth muscle hyper-responsiveness, eosinophil recruitment, and IgE synthesis. Mast cells release proteases that damage airway epithelium and contribute to airway remodeling. Tumor necrosis factor alpha (TNF-α) is a potent bronchoconstrictor produced by mast cells. In chronic asthma there is evidence of persistent mast cell activation resulting in continuous mediator release, cytokine production, and amplification of airway inflammation.18 The presence of mast cells in airway smooth muscle contributes to propagation of the inflammatory response, persistent bronchoconstriction, and over time, airway smooth muscle hypertrophy that plays a role in airway remodeling.19,20 Eosinophils are major effector cells in asthma, and their presence is evidence of the allergic nature of this disease. Increased numbers of eosinophils are found in the airways of most but not all asthmatics.21 Eosinophils contain granules that release inflammatory mediators, including major basic protein, eosinophil cationic protein, and eosinophil peroxidase. Major basic protein is a potent bronchoconstrictor. Eosinophil cationic protein increases airway mucous production and causes histamine release from mast cells. Leukotrienes (LTs) produced by eosinophils are bronchoconstrictors and promote the secretion of thick viscid mucus, leading to airway plugging, LTs also enhance airway vascular permeability, leading to airway edema. Platelet-activating factor, superoxides, and free radicals produced by eosinophils also cause bronchospasm and bronchial tissue injury. Eosinophilic mediators damage and cause desquamation of the airway epithelium, expose nerve endings, provide submucosal access for inflammatory cells and mediators, and negate epithelial cell regulation of the inflammatory process. Although eosinophils appear to be important in airway allergic and inflammatory processes, eosinophilia is not universal in asthmatics. Confounding the role of the eosinophil are studies with anti–IL-5 that significantly reduced sputum and blood eosinophilia but did not significantly affect asthma control.22,23 Clinically, eosinophils are important biomarkers in predicting response to corticosteroid therapy. Steroid-naïve asthmatics with sputum eosinophilia have a good response to inhaled corticosteroid (ICS) therapy, whereas those without sputum eosinophilia do not.24–26 Suppressing sputum eosinophilia with oral or inhaled steroids reduces the number of acute exacerbations in patients with severe asthma.17,27 Mediator and cytokine release from mast cells, eosinophils, and other airway inflammatory cells (macrophages, lymphocytes, basophils, neutrophils) contributes to prolonged bronchial smooth muscle spasm, edema, and mucous production (the late asthmatic response). Ongoing synthesis and release of cytokines, prostaglandins (PGs), and LTs (Fig. 73-4) are responsible for propagation and intensification of airway inflammation and disruption of the airway epithelial border.28 Airway epithelial cells are more than a passive barrier and are recognized to produce proinflammatory mediators. Tight junctions between airway epithelial cells are altered in asthma, which may allow access of allergens, microbes, and viruses into the airway submucosa. Inflammatory cells, mediators, and airway viruses can directly damage the epithelial barrier and stimulate airway epithelial cells to produce inflammatory mediators. Abnormal repair processes may further airway inflammation and obstruction and contribute to airway remodeling.29,30 Nitric oxide (NO) produced by airway epithelial cells in the large and small airways and alveoli is a reflection of ongoing airway inflammation. Measurements of fractional exhaled nitric oxide (FENO) may prove useful for monitoring the response to asthma therapy.31 Airway remodeling refers to the persistent structural changes in airways seen in patients with asthma and is caused by the presence of repetitive or chronic airway inflammation. Microscopic remodeling features include epithelial thickening, subepithelial fibrosis, mucous gland metaplasia, increases in airway smooth muscle, angiogenesis, and loss of cartilage integrity.32 Airway remodeling occurs very early in asthma (childhood) and may precede clinical symptoms.14,33 Remodeling features are prominent in patients with severe asthma.34 Basement membrane thickening may be protective by preventing inflammatory cells and proteins from entering the airway submucosa through a damaged epithelium; simultaneously, this process may be counterproductive by reducing the elasticity of the small airways. Airway remodeling may explain the resistance to therapy observed in patients with prolonged asthma histories and the decline in pulmonary function noted with age. Airway remodeling induced by chronic inflammation may lead to the development of chronic irreversible airflow limitation and increased asthma mortality. Aspirin-exacerbated respiratory disease (AERD) was first described more than 100 years ago. Clinically, AERD includes the tetrad of nasal polyps, eosinophilic sinusitis, asthma, and sensitivity to cyclooxygenase (COX)-1 inhibitor drugs (e.g., aspirin).The prevalence of AERD is 21% in adults with asthma and 5% in children with asthma.35 Ethnic and familial associations are rare, and it occurs more frequently in females.36 Nonsteroidal anti-inflammatory drugs (NSAIDs) also precipitate AERD. AERD is a common precipitant of life-threatening asthma; one survey notes that 25% of asthmatics who require mechanical ventilation have AERD.37 Clinically, most patients with AERD develop symptoms in the third decade (average age of onset is 34 years), frequently after a viral respiratory illness. A previous history of asthma or allergic rhinitis may be noted, but many patients have no prior respiratory disease. Chronic rhinitis, nasal polyps, and anosmia develop and are often followed by chronic pansinusitis. Bronchial asthma and sensitivity to aspirin (acetylsalicylic acid [ASA]) then results. After ingestion of aspirin or a nonsteroidal drug, acute asthma symptoms occur within 3 hours, usually accompanied by profuse rhinorrhea, conjunctival injection, periorbital edema, and occasionally a scarlet flushing of the head and neck.38 The pathogenesis of AERD is detailed in Figure 73-5. ASA inhibits COX,39 of which two isoforms have been identified. COX-1 produces PGs that are involved in normal physiologic maintenance of renal function, gastric mucosal integrity and hemostasis, and inflammatory states. COX-2 is not expressed in normal physiologic circumstances but produces PGs only in response to inflammatory stimuli. COX is necessary for the production of PGE2 in mast cells and eosinophils. PGE2 has a stabilizing effect on these cells via blockade of the enzyme 5-lipoxygenase and the subsequent production of proinflammatory LTs (LTC4, LTD4, and LTE4). AERD occurs as a result of ASA inhibition of COX and a decrease in the production of PGE2. The end results are an increase in production of LTs, some of which are potent bronchoconstrictors, and inflammatory mediator release from mast cells and eosinophils. Most patients with AERD benefit from anti-LT treatment. These agents block synthesis of LTs (e.g., zileuton) or block specific LT receptors (e.g., zafirlukast, montelukast). Desensitization with slowly increased doses of oral aspirin may be used when aspirin avoidance is not possible (e.g., in patients with cardiovascular disease).39 COX-2 inhibitors have the advantage of inhibiting inflammation without renal, gastrointestinal, or hematologic side effects. AERD is not reported after administration of COX-2 inhibitors. These agents provide a potentially safe alternative for treatment of inflammatory conditions in patients with AERD.39,40 Acetaminophen is a poor COX inhibitor. Most patients with AERD can tolerate up to 500 mg of acetaminophen safely, but 28 to 34% experience mild respiratory reactions when administered 1000 to 1500 mg. Reactions to acetaminophen tend to be milder than those to NSAIDs.39 Exercise-induced asthma (EIA) has been recognized since the first Olympic Games. It occurs in 5 to 20% of the general population, 30 to 70% of elite winter and summer endurance athletes, and up to 90% of patients with persistent asthma.41 Atopy is strongly associated with EIA, and up to 40% of patients with allergic rhinitis have EIA. EIA may be another reflection of chronic airway inflammation. Clinically, EIA is usually preceded by 3 to 8 minutes of exercise. Peak symptoms usually occur 8 to 15 minutes after exercise is complete and then begin to remit spontaneously; recovery occurs within 60 minutes. The cause of EIA remains unclear. In the “osmotic” hypothesis, airway cooling leads to mucosal drying and increased surface osmolality that causes mast cell degranulation and release of inflammatory mediators. The “thermal” hypothesis suggests that airway cooling during exercise followed by rapid rewarming after exercise causes airway vascular congestion and increased permeability, resulting in airway edema and obstruction. Another exercise-specific factor is autonomic deregulation associated with prolonged high-intensity physical training. The predominantly parasympathetic drive of athletes (evidenced by low heart rates) may also increase bronchomotor tone and increase the risk of EIA.41 Prophylaxis for EIA with warm-up and a short-acting inhaled beta-agonist is the therapy of choice. Pretreatment with cromolyn, LT antagonists (montelukast), and inhaled parasympatholytics is also effective.42,43 Breathing through the nose may allow warming and humidification of cool dry air during exercise. Long-acting beta-agonists are usually effective, but tachyphylaxis and loss of efficacy may occur if these agents are used regularly.44 Menstruation-associated asthma affects up to 40% of asthmatic women yet receives little emphasis in asthma treatment guidelines.1 The ratio of female-to-male asthma prevalence increases dramatically after puberty, and health care for asthma increases in the perimenstrual phase. Perimenstrual reductions in peak expiratory flow rates (PEFRs) of 35 to 80% are reported. Fluctuations in estrogen and progesterone levels are postulated as causal factors.45,46 Estradiol inhibits eosinophil degranulation and suppresses LT activity; estrogen withdrawal in the luteal phase may enhance these actions. Progesterone may also have bronchodilator and anti-inflammatory activity, and the rapid decline in progesterone levels before menstruation may contribute to increased bronchospasm. Beneficial therapies for perimenstrual asthma include LT antagonists, long-acting beta-agonists, and oral contraceptives.46,47 Psychological factors may precipitate bronchospasm. Panic disorder, generalized anxiety disorder, and mood disorders are more common in asthmatics than the general population. An association between asthma, anxiety, and depression is noted. The mechanisms of bronchospasm associated with chronic stress may be related to the cholinergic nervous system or the hypothalamus-pituitary-adrenal axis, resulting in decreased cortisol secretion.48,49 Compliance may be adversely affected by psychological factors. Relaxation as a therapy for asthma has inconsistent effects; hypnosis may be beneficial.50 It is likely that asthma is not a single disease but a syndrome with various phenotypes. The natural history of asthma is highly variable. Most asthma begins in childhood and resolves with age. Remissions of variable duration may occur, whereas others experience progressive severe disease. Many aspects of asthma (e.g., atopy susceptibility, IgE levels, degree of bronchial hyper-reactivity, asthma severity) have identifiable genetic associations. Family history of both atopy and asthma are associated with lower rates of remission. Environmental influences (e.g., allergens, pollutants, tobacco, and occupational exposures) are associated with asthma, and the interaction of genetic variability and environmental factors may allow prediction of future disease risk, expression, and severity.51,52 The pathology of the asthmatic airway reflects the inflammatory process described earlier. Mucous gland hyperplasia and viscous mucous plugs in the smaller airways are present. Airway secretions reveal increased numbers of inflammatory cells. Airway epithelial damage and remodeling are evident. In contrast to patients with mild to moderate asthma, patients with acute severe asthma have extensive mucous hyperplasia, subepithelial thickening, and infiltration of inflammatory cells (particularly eosinophils).53 Necropsies of patients with status asthmaticus reveal grossly inflated lungs that may fail to collapse on opening of the pleural cavities. Histologic examination reveals luminal plugs consisting of inflammatory cells, desquamated epithelial cells, and mucus. Marked thickening of the airway basement membrane, submucosal inflammatory cells, increased deposition of connective tissue, mucous gland hyperplasia, and hypertrophy of airway smooth muscle are also observed. Reports of slow-onset asthma fatalities reveal greater bronchial eosinophilia and basement membrane thickening when compared with rapid-onset fatal asthma.54,55 Reports of rapid-onset fatal asthma describe less mucus in the airway lumens, suggesting that terminal events may be dominated by bronchoconstriction without excessive luminal plugging; however, an alternative view is that “dry airways” are more reflective of postmortem tissue processing.55 National and International Guidelines for the Diagnosis and Management of Asthma In response to the increasing prevalence of asthma, many countries publish guidelines. Some of these, including NAEPP (National Institutes of Health [NIH]) Expert Panel Report 3 (EPR-3), devote specific portions to the management of acute exacerbations of asthma.1 A task force of physicians representing the American Academy of Asthma, Allergy and Immunology, the American Academy of Emergency Medicine, and the American Thoracic Society (ATS) have summarized the aspects of the EPR-3 that are relevant for the ED and have developed evidence-based recommendations for acute care gaps in the NIH national guidelines. These include (1) use of noninvasive ventilation, (2) use of intubation and mechanical ventilation, (3) appropriate discharge medications, and (4) techniques for ensuring proper follow-up after an ED visit. These additional acute care recommendations were simultaneously published in 2009 in supplemental issues of Journal of Allergy and Clinical Immunology (volume 124, issue 2), Journal of Emergency Medicine (volume 37, issue 2), and Proceedings of the American Thoracic Society (volume 6, issue 4). The EPR-3 and task force recommendations are graded from levels A through D based on the strength of the scientific evidence. ED concordance with the EPR-3 treatment recommendations is reported as moderate, with significant regional variations in quality of care.56 In addition, ED overcrowding is associated with longer lengths of stay for acute asthma exacerbations.57 Although increased airway resistance, diminished flow rates, and increased bronchial hyperactivity are contributing factors, asthmatic patients who come to the ED with nocturnal asthma attacks have disease severity similar to that of other asthmatics. Up to 40% of asthmatic women experience premenstrual worsening of symptoms, which peak 2 to 3 days before menses and are associated with more severe disease; ED visits increase during the preovulatory and perimenstrual intervals.58 There are interindividual differences in the dyspnea perceived by asthmatic subjects for the same level of airway narrowing. This difference results in poor correlation of symptoms with airway obstruction as determined by pulmonary function testing (PFT), both chronically and on presentation to the ED. Patients with a blunted perception of dyspnea (the “poor perceivers”) have more ED visits, hospitalizations, and near-fatal and fatal asthma attacks.59 Many asthmatics report symptoms of gastroesophageal reflux that may cause airway narrowing through a vagally mediated pathway or microaspiration. Proton pump inhibitor therapy decreases asthma symptoms in these patients. Approximately 80% of patients with asthma have symptoms of rhinitis. Approximately 5 to 15% of patients with perennial rhinitis have asthma, and control of sinonasal inflammation can lead to asthma improvement. Overweight (body mass index [BMI] of 25 kg/m2 or higher) asthmatics have poorer asthma control, higher admission rates, and a greater risk of complications, possibly secondary to a difference in the perception of dyspnea or in response to asthma controller agents.60,61 In addition, obese patients have different bronchial and systemic inflammatory characteristics and a specific pattern of pulmonary function changes, suggesting a different asthma phenotype.62 One third of patients who come to the ED with acute asthma are current cigarette smokers, and these patients have poorer asthma control and greater acute care needs than lifelong nonsmokers or former smokers.63,64 African Americans with acute asthma have lower flow rates and more potentially life-threatening episodes than whites, but they respond equally well to albuterol.65 There are no racial disparities in inpatient asthma care when comparing whites, blacks, and Hispanics.66 Slow-onset asthma with progressive deterioration over a period of at least 6 hours (usually days) occurs in over 80% of cases. This type has a female predominance, is triggered by upper respiratory tract infections, and has an airflow inflammation mechanism that results in a slower response to treatment. Sudden-onset asthma with rapid deterioration in less than 6 hours occurs in less than 20% of cases. This type has a male predominance, is triggered by respiratory allergens, exercise, and psychosocial stress, and has a bronchospastic cause resulting in more severe airway obstruction with a faster response to therapy.67 Risk factors for death from asthma are important to determine and are listed in Box 73-1.1,68–71 In urban areas, heroin and cocaine abuse are commonly associated with life-threatening asthma requiring noninvasive positive-pressure ventilation (NIPPV) or intubation in the ED.70,71 The brief history pertinent to the current exacerbation should include onset and possible triggers, severity of symptoms especially as compared with previous exacerbations, and other comorbidities (especially those that may be worsened by systemic corticosteroids such as diabetes, peptic ulcer, hypertension, and psychosis). In addition, all current asthma medications should be noted, including times and amounts recently used, and any potential asthma exacerbators such as aspirin or NSAIDs, beta-blockers (including topical agents used for glaucoma), and angiotensin-converting enzyme inhibitors. Cardioselective and nonselective beta-blocker use increases hospitalizations and ED visits.72 The severity of airflow obstruction cannot be accurately assessed from symptoms and physical examination alone.73 Physicians initially tend to underestimate the degree of airway obstruction in acute asthma. Therefore routine PFT should be part of ED assessment and monitoring. The forced expiratory volume in 1 second from maximal inspiration (FEV1) or the PEFR in liters per second, starting with fully inflated lungs and sustained for at least 10 msec, may be used. Both measurements require the patient’s cooperation for maximal effort and are effort dependent. Whenever possible, the best of three consecutive values should be recorded. Any patient not able to perform a pulmonary function study should be considered to have severe airway obstruction. Although FEV1 can be adequately measured in most acutely ill asthmatics to meet modified ATS performance goals,73 most assessments in the ED use single-patient-use portable peak flow meters because PEFR is easier to measure. The identical device should be used to assess then reassess an individual patient, and different portable meters should not be used interchangeably. Lastly, the FEV1 and PEFR measurements are not interchangeable in assessing acute airway obstruction, which is not addressed in all management guidelines. Although absolute PFT measurements can be used, percentage of predicted performance (% predicted) values are preferable to account for age (now to age 85), sex, and height. ABG sampling should be limited to a subset of patients with predicted PFT values of less than 30%, whose clinical course is perplexing, and for whom capnography is not available. Occasionally, despite improvement in PFT values with bronchodilator therapy, some patients have a transient fall in the partial pressure of oxygen in arterial blood (PaO2) secondary to pulmonary vasodilatation and worsening ventilation-perfusion mismatch.74 The assessment of ventilation may be simplified because there is a high concordance between end-tidal partial pressure of CO2 (PCO2) measured by capnography and the PaCO2 obtained with ABG measurements.75 Also, capnographic waveform analysis can indicate improvements in airway diameter in acute asthma and has the advantages of being effort independent and providing continuous monitoring.76 Noninvasive monitoring of bronchial inflammation may customize the ED assessment of acute asthma. This may include measurement of biologic biomarkers, such as cytokine profiles in the blood, evaluation of LTE4 in the urine, and the monitoring of exhaled pentane, hydrogen peroxide, NO, or carbon monoxide levels. Of these measurements, exhaled NO, a marker of airway inflammation, shows the most promise, as studies show that FENO can be measured in the ED with good reproducibility, and levels measured after 6 hours of care are associated with better asthma control after discharge.77,78 The severity of airflow obstruction cannot be accurately judged by patients’ symptoms, physical examination findings, and laboratory test results. Serial measurements of airflow obstruction (FEV1 or PEFR) are key components of disease assessment and response to therapy (Table 73-1). Table 73-1 Objective Findings in Asthma Assessment
Asthma
Perspective
Epidemiology
Principles of Disease
Miscellaneous Situations
Genetics and Asthma
Pathology
Clinical Features
Symptoms
Historical Components
Diagnostic Strategies
Arterial Blood Gas Analysis
Future Monitoring Strategies
Assessment Summary
FACTOR
SEVERE ASTHMA (FEV1 <1.0 L)
Pulse rate (beats/min)
≥120 but may be less with equally severe asthma
Respiratory rate (breaths/min)
≥40 but most are >20, therefore nondiscriminating
Pulsus paradoxus (mm Hg)
≥10 but may be absent with equally severe asthma in 50% of cases
Pulse rate ≥120, respiratory rate ≥20, pulsus paradoxus ≥10
If all three abnormal, 90% with severe asthma, but only 40% with FEV1 <1.0 L have all three abnormal
Use of accessory muscles of respiration
If present, may indicate severe asthma; if absent, may have equally severe asthma in 50% of cases
ABG analysis (mm Hg)
PaO2 ≤60 or PaCO2 ≥42 indicates severe asthma; all other values difficult to interpret unless PEFR or FEV1 known
Pulmonary function studies
PEFR and FEV1 measure directly the degree of airflow obstruction; most useful in assessing severity and guiding treatment decisions

Full access? Get Clinical Tree


Asthma
Only gold members can continue reading. Log In or Register to continue




