CHAPTER 25 Anesthesia for Plastic Surgery
For the pediatric plastic surgeon, a significant portion of reconstructive surgery involves pathologies of the craniofacial skeleton and soft tissue, and these may be of significant concern for the pediatric anesthesiologist. In this chapter, the pathologies that result in surgery are set in the framework established by Whitaker and associates in 1979. The anesthesiologist who cares for these children will find it easier to organize the varied syndromes and sequences when they are structured in this manner.
Craniofacial surgery
Craniofacial anomalies are characterized by congenital or acquired deformities of the cranial or facial skeleton. Although rare, they comprise a diverse group of defects, and the goal of surgical intervention is to restore both form and function. The classification of craniofacial anomalies is difficult because of their variability, rarity, and degree of severity, and often because of our lack of understanding of their etiology and pathogenesis. The Committee on Nomenclature and Classification of Craniofacial Anomalies of the American Cleft Palate Association has proposed the following categories: synostosis, clefts, hypoplasia, hyperplasia, and unclassified (Whitaker and Bartlett, 1990).
Craniosynostosis
Craniosynostosis is defined as premature closure of one or more of the cranial sutures. It is a relatively common defect, occurring in one in 2000 to 2500 live births (Posnick, 2000). It can result in abnormalities in the size and shape of the calvarium, cranial base, and orbits, and can cause dental occlusion, and thus it constitutes a diverse group of deformities. Craniosynostosis is not only a cosmetic issue, for it can also affect brain growth, intracranial pressure (ICP), and vision, resulting in developmental delay and vision impairment (Slater et al., 2008).
Premature fusion of a cranial suture results in an abnormal head shape. Growth of the skull perpendicular to the suture is impaired. Compensatory growth parallel to the suture creates the characteristic abnormal skull shape. The shape of the skull defines craniosynostosis. Figure 25-1 shows the different types of abnormal skull shapes and the corresponding synostotic sutures.
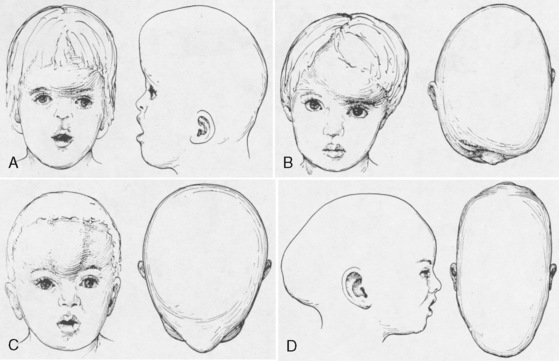
Craniosynostosis can occur by itself (simple) or as a major component of a syndrome (complex, or syndromic). Over 100 craniosynostosis syndromes have been described, but six are more common than the others (Slater et al., 2008). These are Apert, Pfeiffer, Saethre-Chotzen, Carpenter’s, Crouzon, and Muenke’s syndromes. Of these, Apert and Crouzon syndromes are the most common. Table 25-1 lists the various syndromes and their associated anomalies and anesthetic concerns. The timing of surgery usually occurs before 1 year of age. The importance of early intervention is related to improved ability of the infant to reossify, the malleable nature of the calvaria during infancy, and the rapid brain growth that occurs during the first year of life (Panchal and Uttchin, 2003). The motivation for and goal of surgical intervention are to reduce ICP, prevent brain injury, and enhance appearance. Repair of syndromic craniosynostosis may be more complicated and appears to be associated with increased blood loss. The etiology of the increased bleeding is unclear, but it might be related to the length of surgery (Fearon and Weinthal, 2002).
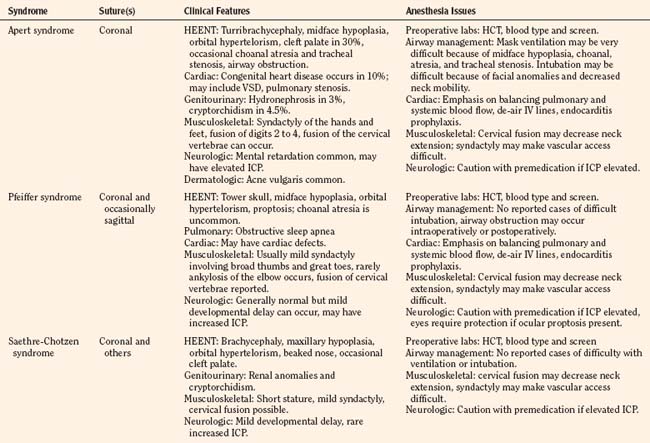
Apert Syndrome
Apert syndrome, also referred to as acrocephalosyndactyly, occurs at a frequency of 1 in 160,000 live births (Cohen et al., 1992). Acrocephalosyndactyly, a defect involving the cranium and the extremities, occurs in Apert, Pfeiffer, Carpenter’s, and Saethre-Chotzen syndromes. The characteristic features of Apert syndrome include turribrachycephaly (high steep flat forehead and occiput), mid-face hypoplasia, and orbital hypertelorism (Fig. 25-2). Cleft palate occurs in approximately 30% of these patients. Choanal atresia and occasionally tracheal stenosis have been reported and can cause airway obstruction. Congenital cardiac disease is one of the more common associated visceral anomalies, occurring in approximately 10% of patients. Genitourinary anomalies (hydronephrosis, cryptorchidism) also occur in 10% of patients with Apert syndrome (Cohen and Kreiborg, 1993). Severe synostosis can result in increased ICP and, if uncorrected, developmental delay. Syndactyly of the hands and feet with the fusion of digits two to four can occur and can make intravenous (IV) access difficult. Kreiborg and coworkers (1992) have reported cervical spine fusion, and they have suggested that cervical spine films prior to anesthesia may help predict difficult intubation. Although infants and children with Apert syndrome are often difficult to intubate, many have been intubated uneventfully. In some cases, suboptimal laryngoscopic views secondary to abnormal anatomy may require flexible fiberoptic intubation. The laryngeal mask airway (LMA) may be a reasonable adjunct in those patients who are difficult to ventilate or intubate. However, to date there are no reported instances of their use in infants and children with Apert syndrome. The clinical features and anesthetic implications of Apert syndrome and the other acrocephalosyndactylies are outlined in Table 25-1. Unlike Apert syndrome, the other acrocephalosyndactylies are not typically associated with difficult airways. However, midface hypoplasia is common in these infants and may cause significant upper airway obstruction intraoperatively and postoperatively (Perkins, 1997).
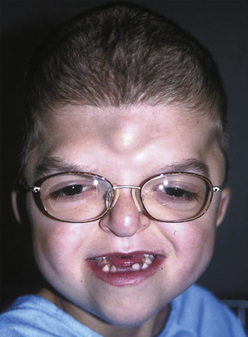
Pfeiffer Syndrome
Pfeiffer syndrome is another example of an acrocephalosyndactyly. The incidence of Pfeiffer syndrome is approximately 1 in 100,000 live births. This syndrome is characterized by bicoronal synostosis, proptosis, midface hypoplasia, and broad thumbs and great toes (Moore et al., 1995). Patients with Pfeiffer syndrome can also present with hydrocephalus, which may contribute to increased intracranial pressure. There are three types of Pfeiffer syndrome, and typically the clinical features, degree of airway obstruction, and mortality rates increase with types 2 and 3 (Moore et al., 1995).
Saethre-Chotzen and Carpenter’s Syndromes
The clinical features of Saethre-Chotzen include brachycephaly, facial asymmetry, low hairline, proptosis, beaked nose, large halluces (great toes), and pectus excavatum. Some patients may have renal anomalies, cryptorchism, developmental delay, and epilepsy. It can be difficult to differentiate it from the other acrocephalosyndactylies because there can be significant clinical variability (Nascimento et al., 2004). Carpenter’s syndrome is the most rare of the syndromic craniosynostosis, with only 45 reported cases since the mid 1990s. As with all of the syndromic craniosynostosis, patients with Carpenter’s syndrome have synostosis, midface hypoplasia, and musculoskeletal deformities. They may also have hypogonadism, developmental delay, and obesity (Idestrand et al., 2009).
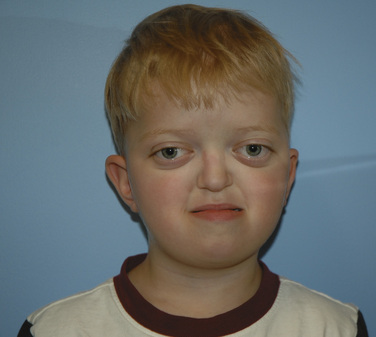
Crouzon Syndrome
Crouzon syndrome, also known as craniofacial dysostosis, is one of the syndromic craniosynostoses. These infants present with craniosynostosis, proptosis, and midface hypoplasia but without visceral or extremity involvement (Fig. 25-3). As in other patients with midface hypoplasia, significant airway obstruction can occur and may require early tracheostomy (Sirotnak et al., 1995). Table 25-1 outlines the main clinical features and anesthetic issues as they relate to patients with Crouzon syndrome. During infancy, patients with Crouzon syndrome may come to the operating room for tracheostomy or cranial vault remodeling.
Surgical Management
Cranial Vault
Surgical correction for craniosynostosis can be performed with an open or an endoscopic procedure. The calvarial vault reconstruction typically involves both plastic surgery (by craniofacial surgeons) and neurosurgery. The surgical approach for an open cranial vault reconstruction is through a bicoronal incision (Fig. 25-4, A). A blocking stitch or clips may be applied to the skin flaps to minimize blood loss. The clips may be more effective in preventing bleeding, but some surgeons have expressed concern about the risk for ischemia of the underlying hair follicles (Ray Harschberger, personal communication, 2009). The scalp flap is dissected off the forehead and mobilized down to expose the superior orbital rim (see Fig. 25-4, B). The calvarium is typically removed by the neurosurgeons in one or several pieces. A bandeau osteotomy is then performed along the lateral temporal bones and the nasion to mobilize the superior orbital rim (see Fig. 25-4, C). Once the osteotomies are complete, the surgical field is protected with moist gauze. The calvarium and the orbital bandeau are sectioned and the pieces are reshaped and replaced in a manner that replicates a normal head shape. The bone is secured with a craniofacial plating system, the scalp flaps are replaced, and the coronal incision is closed.
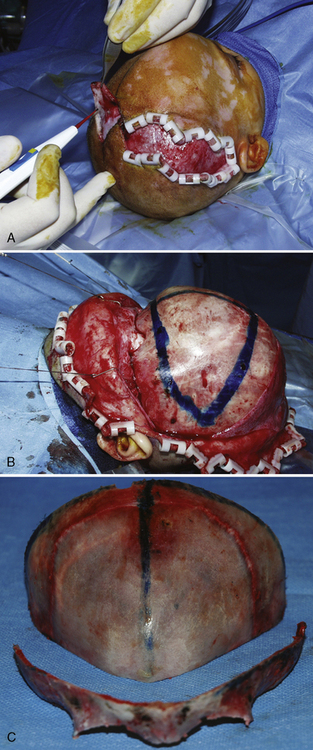
Strip Craniectomy with Helmet
The endoscopic approach is less invasive and has been reported to cause less blood loss (Jimenez et al., 2002). This procedure is more commonly used for sagittal synostosis, although it has been described for the repair of other single suture and even multiple-suture synostosis. The surgical approach is through smaller incisions. As with open approaches, significant blood loss can occur if the sagittal sinus is entered. Jiminez reported a small percentage of patients who required transfusions, and most were discharged on the first postoperative day. Unlike those who had an open calvarial reconstruction, these patients do require helmet therapy after this repair.
Midface Advancement
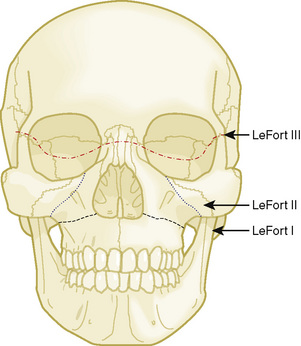
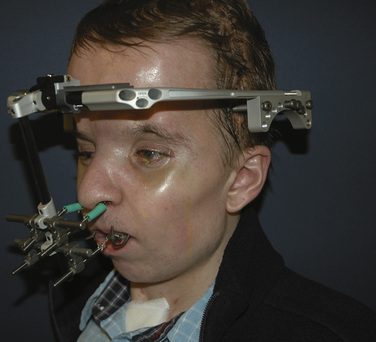
Midface advancements are performed to correct midface hypo-plasia. The different types of advancements include the Le Fort I maxillary advancement, the Le Fort III maxillary and upper face advancement (Fig. 25-5), and the monobloc advancement, which includes the maxilla, upper face, orbits, and forehead. The Le Fort I osteotomy is typically performed for patients with midface hypoplasia secondary to cleft lip and palate. The Le Fort III and monobloc osteotomies are typically performed for patients with midface hypoplasia secondary to syndromic craniosynostosis (Apert, Pfeiffer, Crouzon syndromes). The surgical approach for the Le Fort III and monobloc osteotomies are through bicoronal, intraoral, and often eyelid incisions. Significant blood loss can occur during the surgical dissection and osteotomies. Once the midface is mobilized, the advancement can be performed immediately with rigid fixation or gradually with internal or external distraction osteogenesis. Distraction osteogenesis became a common in craniofacial surgery in 1997, and the advantages include less morbidity, more long-term stability, and improved aesthetic outcome (Fearon, 2001; Shetye et al., 2007). The external distraction device has a frame that is anchored to the skull, with a distraction bar positioned perpendicular to the face. The distraction bar is anchored to the newly mobilized midface with wires (Fig. 25-6). Once in place, the midface may be distracted at a rate of 1 to 2 mm a day.
Anesthesia Management
The anesthetic management of infants with craniosynostosis begins with a complete preoperative evaluation. The history should define the cranial sutures involved, the planned surgical procedure, and previous craniofacial reconstruction, and it should identify any associated syndrome. Infants and children with syndromes may have difficult airways, other organ involvement, and more complicated surgical repair with more bleeding. Associated anomalies that can present a challenge to the anesthesiologist include facial and airway features that make mask ventilation and intubation difficult. Airway pathology can also cause obstruction, and some of these children have obstructive sleep apnea (OSA) (Mixter et al., 1990; Pijpers et al., 2004).
Children with OSA may present with daytime somnolence, enuresis, behavioral changes, and snoring. As many as 40% to 50% of infants and children with syndromic craniosynostosis have clinical features of OSA. This compares with the 0.2% to 0.7% incidence of OSA in the general pediatric population (Lo and Chen, 1999; Pijpers et al., 2004). The most common treatment for OSA is adenotonsillectomy. If this fails to reduce the symptoms, the sequence of recommended steps in this population includes nasal continuous positive airway pressure (CPAP), Le Fort III osteotomy with midface advancement, and as a last resort, tracheostomy. Several studies have demonstrated a reduction in the symptoms of OSA after midface advancement. In one study by Nelson and colleagues (2008), all of the patients reported a decrease in snoring, and five of the six patients with a tracheostomy could be decannulated.
A history of fatigue (or sweating with feedings), cyanosis, and syncope are suggestive of an underlying cardiac anomaly. Cardiac pathology is associated with some of the syndromes (e.g., Apert, Pfeiffer, Carpenter’s). Congenital heart disease is most common in Apert syndrome, and the most common cardiac defect is ventricular septal defect (Hidestrand et al., 2009). Infants and children with clinical signs or symptoms suggestive of heart disease or a heart murmur should be preoperatively evaluated by a pediatric cardiologist.
Some infants and children with craniosynostosis may have increased ICP. The incidence of increased ICP in nonsyndromic craniosynostosis varies from 8% to 47%, depending on the number of sutures involved (Arnaud et al., 1995; Renier et al., 2000; Mathijssen et al., 2006). The incidence appears to be higher in syndromic craniosynostosis, with approximately 50% having funduscopic evidence of papilledema (Bannink et al., 2008). This may manifest as headaches, vomiting, and somnolence. However, infants and children with chronically elevated ICP may be asymptomatic. Elevations in ICP manifest as papilledema on ophthalmologic examination, so all patients with craniosynostosis should have a funduscopic examination by an ophthalmologist.
Airway
Airway management in these patients can be very challenging during attempts at ventilation, intubation, or both. Fortunately, difficult airways are not common. However, the incidence is higher in those patients with syndromes and in those patients who have had previous reconstruction. Successful techniques described for infants and children include using the Bullard laryngoscope, laryngeal mask airway, flexible fiberoptic scope, and retrograde intubation (Cooper and Murray-Wilson, 1987; Blanco et al., 2001; Brown et al., 1993, 2004) (see Chapter 10, Equipment, and Chapter 12, Airway Management). A combination of techniques may be required to secure the airway. For example, the LMA has been used to facilitate the passage of the fiberoptic scope and endotracheal tube (Inada et al., 1995) (Fig. 25-7). Although those authors report this technique in a patient with Treacher Collins syndrome, it could be used for any craniofacial patient with a difficult airway. An endotracheal tube and LMA size chart helps in choosing the appropriate equipment (Table 25-2, and see also Table 10-3 in Chapter 10, Equipment). Cuffed endotracheal tubes may be more challenging, because the cuff on the endotracheal tube may not fit through the smaller LMAs. However, the air-Q (Trudell Medical Marketing, London, Ontario, Canada) has its short stalk and detachable adapter that make it easy to pass a cuffed endotracheal tube with the pilot balloon. If an uncuffed endotracheal tube is used, it can be secured while removing the LMA by telescoping a smaller endotracheal tube through the top of the larger tube (see Fig. 25-7, B). When passing an endotracheal tube through an LMA that is a size 3 or smaller, an uncuffed endotracheal tube is easier to use, but it runs the risk for creating a large air leak.
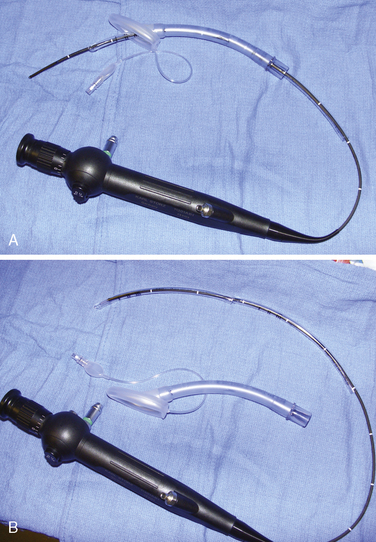
TABLE 25-2 Equipment Sizes for Airway Management
| Laryngeal Mask Airway (size) | Endotracheal Tube (mm) |
| 1.0 | 3.0 uncuffed |
| 1.5 | 4.0 uncuffed |
| 2 | 4.5-5.0 uncuffed |
| 2.5 | 5.5 uncuffed |
| 3 | 6.0 uncuffed |
| 4 | 6.0 cuffed |
Patients having Le Fort osteotomies require a nasal or an oral intubation depending on the type of Le Fort. Those having a Le Fort I osteotomy (for clefts) will need a nasal endotracheal tube so the maxillae and the mandible can be aligned properly. Nasal endotracheal tubes have been damaged during Le Fort osteotomies, resulting in difficulty with ventilation (Bidgoli et al., 1999), so care must be taken by the surgeons when mobilizing the maxillae. Patients having a Le Fort III osteotomy for midface distraction can be intubated orally with an oral RAE tube.
Some infants with craniofacial anomalies require tracheostomy because of significant upper airway obstruction (Perkins, 1997; Sculerati et al., 1998). Adequate preparation entails having all the necessary equipment available and having personnel who are trained and experienced in the use these airway instruments. It may also mean having a pediatric otorhinolaryngologist immediately available.
Blood Conservation and Transfusion Medicine
Craniofacial procedures are often long, exposing infants to the risks of hypovolemia, hypothermia, blood loss, and venous air emboli. The craniofacial procedures typically performed during the first year of life include cranial vault remodeling (including fronto-orbital, posterior, and total vault advancement) and strip craniectomy. One of the most pressing concerns related to anesthesia care in craniofacial surgery is the management of intraoperative bleeding. The cranial procedures can involve significant blood loss because of the duration of the procedure, the many exposed skin and bone surfaces, and the rare complication of entering large vessels such as the sagittal sinus. Blood loss during these procedures has remained an issue since Whitaker and colleagues’ description of perioperative blood loss in 1979. Blood loss is often as high as half to one blood volume, and nearly 90% to 100% of the infants undergoing these procedures may require a blood transfusion (Faberowski et al., 1999; Tuncbilek et al., 2005; Stricker et al., 2010). Even the strip craniectomy, which is typically performed to correct nonsyndromic isolated sagittal synostosis and results in less blood loss, can produce significant hemorrhage. In a recent study by White and others (2009), predictors of blood loss during craniofacial surgery included surgery time greater than 5 hours, age less than 18 months, the presence of multiple-suture craniosynostosis, and syndromic craniosynostosis. Infants undergoing strip craniectomy usually experience less blood loss, but this population may be at increased risk because they come to the operating room at the nadir of their physiologic anemia (i.e., at 2 to 3 months of age). Preparation for these procedures requires a baseline hematocrit, and blood typing and cross-match. Some centers also obtain coagulation studies. Adequate IV access needs to be obtained for resuscitation. In an infant, at least two large-bore (22- to 20-gauge) peripheral IV catheters should be placed to provide adequate access. Arterial pressure monitoring is recommended for beat-to-beat analysis of blood pressure and intravascular volume status, as well as for blood gas monitoring. A central venous catheter may be placed for central venous pressure monitoring, or in patients with difficult IV access.
Preoperative blood donation has been described in children as small as 8 kg (Mayer et al., 1996), but this technique has significant limitations in the infant. Their young age, small size, and lower hematocrit may make blood collection more challenging and less feasible. Acute normovolemic hemodilution has been described in older children and adolescents and may be effective. Hemodilution is relatively contraindicated in the infant less than 6 months old. The normal physiologic advantage with hemodilution (increased preload, increased stroke volume, and decreased systemic vascular resistance, increased tissue oxygenation) may be lost in the infant because of fetal hemoglobin, which more avidly binds oxygen, and a naturally less compliant myocardium.
Because most infants presenting for cranial vault reconstruction have a normal physiologic anemia, erythropoietin was proposed as a therapy to address this concern. Preoperatively, recombinant erythropoietin decreases the transfusion requirements in infants having craniosynostosis repair. Fearon and Weinthal (2002) reported the dosage of erythropoietin as 600 units/kg given subcutaneously once per week along with oral iron supplementation. Erythropoietin was started 3 to 4 weeks before surgery, and the incidence of blood transfusions in infants having craniosynostosis repair decreased from 93% to 57%.
In March of 2007, the U.S. Food and Drug Administration placed a black-box warning on synthetic erythropoietin because of the concern of increased death, deep venous thrombosis, and cancer spread in adult patients receiving synthetic Epogen (Singh et al., 2006; see also www.fda.gov/for consumers consumers). These concerns have occurred only in the adult population, but some centers have stopped using synthetic erythropoietin for craniofacial surgery. A multicenter retrospective review of 396 pediatric craniofacial patients receiving erythropoietin did not reveal an increase in perioperative complications, specifically death or deep venous thrombosis (Naran et al., 2010).
Antifibrinolytic therapy has been described in pediatric surgery, but the data for craniofacial surgery are limited. Aprotinin, a serine protease inhibitor, decreases perioperative blood loss and transfusion requirements during craniofacial procedures. In a prospective, randomized, and blinded placebo-controlled study evaluating the effect of aprotinin in infants and children having cranial vault remodeling and frontal orbital advancements, D’Errico and colleagues (2003) noted a reduction in the amount of packed red blood cells being transfused intraoperatively and postoperatively in those receiving aprotinin. However, aprotinin did not reduce the number of patients requiring transfusion. Although no adverse events were reported, the option of aprotinin has been curtailed with its removal from the market. Tranexamic acid and aminocaproic acid are two antifibrinolytics that remain available for potential pediatric use. The data to guide the use of tranexamic acid and aminocaproic acid in this population are limited, but a small study by Durán de la Fuente and colleagues (2003) demonstrated a reduction in estimated blood loss and transfusion requirements. The tranexamic acid was not infused continuously but instead was administered every 8 hours at a dosage of 15 mg/kg. Table 25-3 shows standard dosage regimens for tranexamic acid and aminocaproic acid.
TABLE 25-3 Dosage Guidelines for Antifibrinolytic Therapy
| Drug | Dosage |
| Aminocaproic acid (Amicar) | Load 100 mg/kg |
| Infuse 10 mg/kg per hr | |
| Tranexamic acid | Load 10-100 mg/kg |
| Infuse 1-10 mg/kg per hr |
In the past, the use of a cell saver was reported as being impractical for small pediatric patients because of the size of the collection reservoir (De Ville, 1997). Recently, the cell-saver reservoirs are available in sizes as small as 25 mL. This technology may reduce the rate of autologous blood transfusion in infants having craniofacial surgery. In fact, the most significant benefit may occur when cell-saver technology is combined with erythropoietin pretreatment. In a prospective analysis evaluating the use of cell saver with a 55-mL pediatric bowl for patients pretreated with erythropoietin, only 30% of those infants having cranial vault remodeling required allogeneic blood (Fearon, 2005). Krajewski and coworkers (2008) in a prospective randomized trial describe a 5% transfusion rate in infants having elective craniosynostosis surgery when both cell saver (25-mL collection reservoir) and erythropoietin were used, compared with a 100% transfusion rate for those not receiving cell-saver techniques or erythropoietin.
Temperature and Positioning
Craniofacial procedures can be very long—sometimes several hours. Complications resulting from long surgical procedures include skin breakdown, neuropathic injury, and hypothermia. Attention must be paid to the initial setup to ensure adequate positioning and padding to minimize these intraoperative injuries. Infants having cranial vault remodeling may be positioned prone, and attention to protecting the face and eyes is important. Patients with midface hypoplasia and proptosis may present a challenge when placed prone, because adequately protecting the face and eyes may be more difficult. The infant is placed on a full-access forced-hot-air blanket to minimize hypothermia, and the surgical site (head) is then isolated from the body using plastic drapes (Fig. 25-8). This not only minimizes convective and radiant heat losses but also prevents conductive heat loss to a bed wet from irrigation and blood. Blood products should be warmed through a fluid warmer before administration (except for platelets). Intravenous fluid warmers may be used, particularly if large volumes of cold allogeneic blood are being administered.
Venous Air Embolism
Venous air embolism (VAE) is a potential complication of craniofacial and neurosurgical procedures (see Chapter 22, Anesthesia for Neurosurgery). VAE can present with hemodynamic instability and can result in death. VAE occurs commonly in pediatric patients having cranial procedures. A prospective study using precordial Doppler in infants and children having craniosynostosis repair detected VAE in 82% of the patients; 31% percent developed hypotension secondary to VAE, but none developed cardiovascular collapse (Faberowski et al., 2000). This is higher than the previously reported incidence of 66% (Harris et al., 1987). Infants may be at increased risk for VAE, because significant hemorrhage during cranial vault remodeling can result in low central venous pressures. In addition, the relatively large size of the infant head may raise the surgical site above the level of the heart, thereby increasing the pressure gradient for air entrainment. Some advocate the placement of central venous catheters to monitor the trend of central venous pressures and minimize the risk for air embolism. However, no data suggest that central venous pressure monitoring decreases the risk for VAE.
Management of VAE begins with preventing hypovolemic states by providing adequate volume resuscitation and using a precordial Doppler for early detection of VAE. The precordial Doppler should be placed over the left or right parasternal border between the third and sixth intercostal spaces (Schubert et al., 2006). Lowering the head of the bed, left lateral positioning, flooding the surgical field with saline, applying bone wax, discontinuing nitrous oxide, and providing inotropic support are all measures that have been used to acutely manage VAE. Pediatric advanced life support with chest compressions and epinephrine is required for patients who develop symptomatic low blood pressure, bradycardia (heart rate < 60 bpm), or pulseless electrical activity.
Postoperative Care
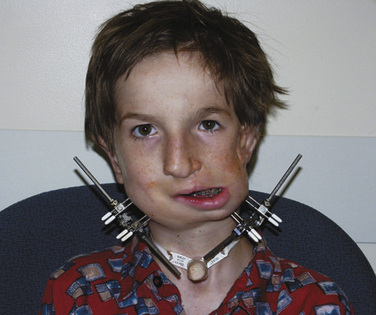
The postoperative treatment of infants having craniofacial surgery depends on coexisting morbidities and the procedure performed. Most infants and children presenting for cranial vault reconstruction or midface advancement can be extubated in the operating room at the completion of surgery. Factors that may preclude extubation include intraoperative complications resulting in hemodynamic instability, known difficult airway, significant tongue swelling, and intranasal cerebrospinal fluid drainage after Le Fort III osteotomy. Infants in whom distractors were placed may have a more difficult airway after extubation (Fig. 25-9). Mask ventilation can be very difficult with mandibular distractors and impossible with midface distractors. Airway equipment, including oral and nasal airways and appropriately sized LMAs, should be available after extubation. Equipment and personnel to remove part of the distractor device are also important in the operating room (Wong et al., 2004).
Infants having cranial vault remodeling and frontal orbital advancements can experience significant blood loss intraoperatively. If these patients are adequately resuscitated and are hemodynamically stable, they can often be extubated in the operating room. Infants with difficult airways or significant airway obstruction, or those who have experienced intraoperative complications, may benefit from delayed extubation in the intensive care unit (or in the operating room) after their condition has stabilized. Ongoing blood loss is common after major craniofacial surgery, and infants may require repeat transfusions in the immediate postoperative setting. Other complications include cerebral edema (Levine et al., 2001), visual changes (Lo et al., 2002), cerebrospinal fluid leak (Fearon, 2003), infection (Fialkov et al., 2001), metabolic acidosis, and transfusion reactions.
Hyponatremia has been reported after major intracranial procedures in pediatric patients. This was originally attributed to syndrome of inappropriate diuretic hormone (SIADH). However, this diagnosis has come into question. Although it is not entirely clear, cerebral salt wasting syndrome (CSW) may be the more likely cause. Both SIADH and CSW manifest with hyponatremia. CSW is differentiated from SIADH by hypovolemia, high urine output, and low or normal antidiuretic hormone levels (Levine et al., 2001) (Table 25-4). The proper diagnosis is important because the two are managed differently. SIADH is treated with free water restriction, whereas CSW is managed with isotonic fluid replacement (normal saline) to cover maintenance requirements and urine losses. Frequent sodium evaluations are important to guide therapy.
TABLE 25-4 Comparison of Syndrome of Inappropriate Antidiuretic Hormone (SIADH) and Cerebral Salt Wasting (CSW)
| Monitoring | SIADH | CSW |
| Central venous pressure | High (>5) | Low (<5) |
| Urine output | Decreased | Increased |
| Urine sodium level | High (>20 mmol/L) | High (>20 mmol/L) |
| Urine osmolality | High | Low or normal |
| Serum osmolality | Low | Low or normal |
| Antidiuretic hormone | High | Normal |
Hypoplasia
Pierre Robin Sequence
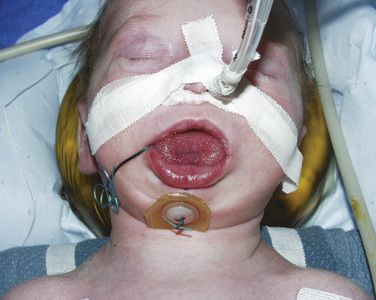
Neonates and infants with PRS will have some degree of airway obstruction. They often also present with significant reflux and feeding difficulties. Mild airway obstruction may require only lateral or prone positioning to relieve the airway obstruction; however, as many as one quarter of infants with PRS have more severe obstruction, necessitating surgical intervention (Bijnen et al., 2009). The optimal surgical intervention is not clear, and there is significant regional variation. Preoperative evaluation by a pediatric plastic surgeon and pediatric otolaryngologist is essential to completely evaluate the airway and identify coexisting airway pathology. The management options include tongue-lip adhesion, mandibular distraction, and tracheostomy. A tongue-lip adhesion involves suturing the inferior portion of the tongue to the lower lip to prevent the tongue from falling to the back of the pharynx and causing obstruction (Fig. 25-10). Once the mucosal flaps from the tongue and the lower lip are approximated, a temporary retention suture with a button is placed through the floor of the mouth and around the mandible to secure the repair. The infant is intubated postoperatively for several days to ensure the success of the adhesion. The goal of the tongue-lip adhesion is to relieve the airway obstruction and improve feeding until the mandible has had a chance to grow. Some evidence suggests that this repair is effective at decreasing airway obstruction and improving feeding (Kirschner et al., 2003). Bijnen and coworkers (2009) found that 73% of their patients had improved airway patency, based on sleep studies, and improved feeding. The tongue-lip adhesion is left intact until the patient is approximately 1 year old. At that time, the mandible has had time to grow and the adhesion can be taken down.
An alternative therapeutic approach involves mandibular distraction. In infants with severe obstruction, mandibular distraction can be performed to rapidly distract the jaw. It can be performed in infants less than 6 months old. Genecov and colleagues (2009) recently described their 10-year experience with mandibular distraction for PRS. Of the 67 patients that had preoperative polysomnography, 65 experienced an improvement. Also, several of the patients who required tracheostomy for PRS could be decannulated after their mandibles were distracted.
Tracheostomy is a final option for those patients who fail conservative treatment, tongue-lip adhesion, or distraction. Some have advocated for early tracheostomy for PRS, but there is evidence that it involves increased morbidity and mortality. Also, once infants receive a tracheostomy, it takes on average 3 years for decannulation to occur (Tomaski et al., 1995).









