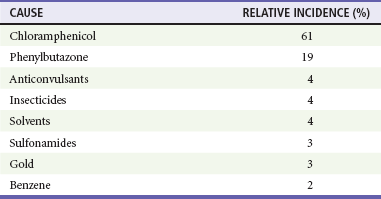
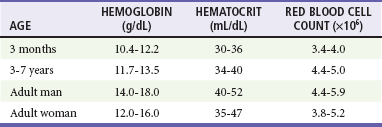
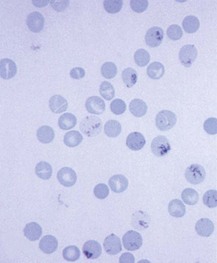
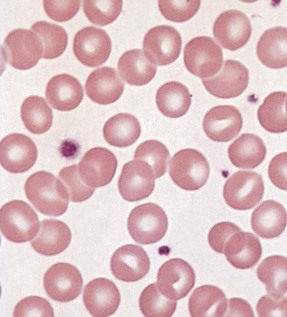
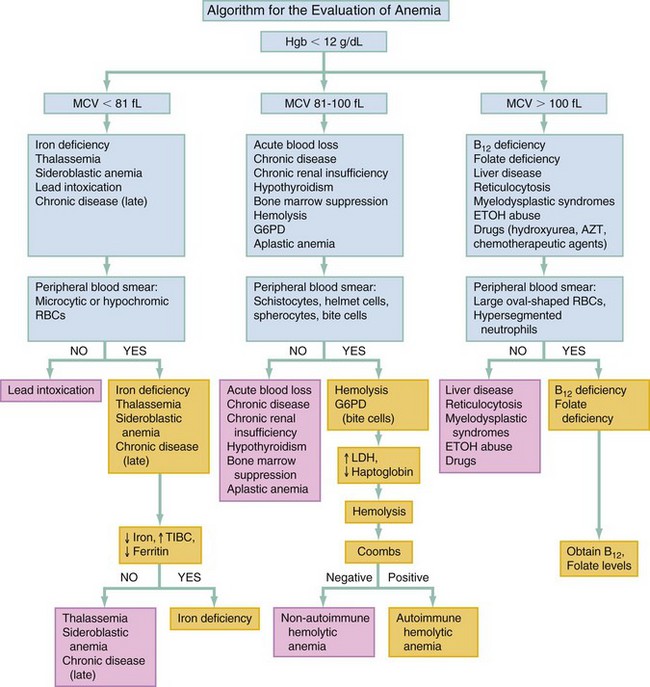
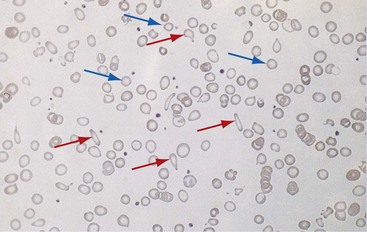
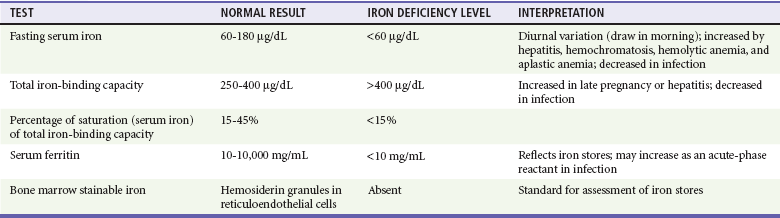
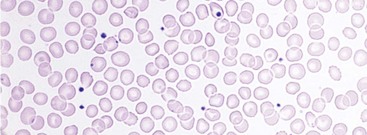
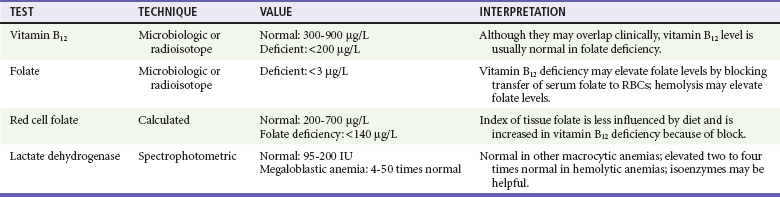
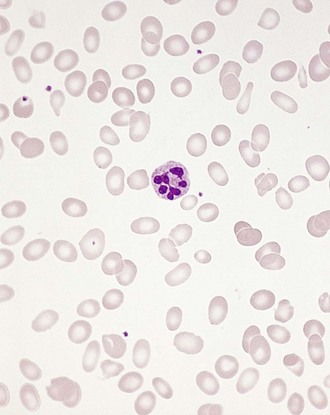
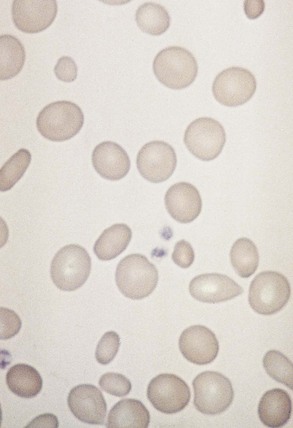
Chapter 121 Anemia is an absolute decrease in the number of circulating red blood cells (RBCs). The diagnosis is made when laboratory measurements fall below accepted normal values (Table 121-1). Bone marrow contains pluripotent stem cells that can differentiate into erythroid, myeloid, megakaryocytic, and lymphoid progenitors. Erythropoietin enhances the growth and differentiation of erythroid progenitors. When the late normoblast extrudes its nucleus, it still contains a ribosomal network, which identifies the reticulocyte (Fig. 121-1). The reticulocyte retains its ribosomal network for approximately 4 days, 3 days of which are spent in bone marrow and 1 day in the peripheral circulation. The RBC matures as the reticulocyte loses its ribosomal network and circulates for 110 to 120 days. The erythrocyte is then removed by macrophages that detect senescent signals. Causes other than blood loss may be responsible for severe anemia of rapid onset. Certain rare hemolytic conditions can cause rapid intravascular destruction of RBCs (Box 121-1). More common are patients with chronic compensated hemolytic anemia (e.g., sickle cell disease), who decompensate with an acute-onset anemia as a result of decreased erythrocyte production triggered by a viral infection. Clinical signs and symptoms include tachycardia, decreased blood pressure, postural hypotension, lightheadedness, increased heart rate, and increased respiratory rate. Complaints of thirst, altered mental status, and decreased urine output may also be present. The patient’s age, concomitant illness, and underlying hematologic, cerebral, and cardiovascular status tremendously influence the clinical findings. Children and young adults may tolerate significant blood loss with unaltered vital signs until a precipitant hypotensive episode occurs. Pediatric patients may become markedly tachycardic, physiologically attempting to maintain cardiac output because their ability to increase stroke volume is limited. Elderly patients commonly have underlying disease states that compromise their ability to compensate for blood loss.1 Pertinent elements of the history and physical examination of patients with acute anemia are listed in Box 121-2. Nonemergent anemias are usually seen in ambulatory patients complaining of fatigue and weakness. Other voiced complaints include irritability, headache, postural dizziness, angina, decreased exercise tolerance, shortness of breath, and decreased libido. When the anemia is of slow onset, the patient may adapt until the hemoglobin is very low. Alternatively, patients with rapid blood loss may experience lightheadedness or syncope even when the measured hemoglobin is not critically low. For patients without evidence of acute bleeding or emergent condition, elements of history and physical examination may help identify the cause (Box 121-3). Most of these patients do not need immediate stabilization and can be further evaluated as outpatients. The initial laboratory evaluation includes a complete blood count with leukocyte differential, reticulocyte count, peripheral smear (Fig. 121-2), and RBC indices, including mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), and mean corpuscular hemoglobin concentration (MCHC). The differential diagnosis of anemia is facilitated by classification of the anemia into one of three groups: decreased RBC production, increased RBC destruction, and blood loss. A complementary approach uses RBC morphology and indices. Figure 121-3 presents an algorithm for the evaluation of anemia. Anemias caused by decreased RBC production have a natural history of insidious onset and an associated decreased reticulocyte count. A subclassification by indices of anemias caused by decreased RBC production is listed in Box 121-5. The RBC indices and morphology manifested in a peripheral smear are useful in securing the diagnosis. The definitive diagnosis is usually made outside the emergency department and may require bone marrow examination. The emergency physician rarely initiates replacement therapy, except in circumstances that require transfusion. Appropriate diagnostic tests may be initiated, but replacement of iron, vitamin B12, or folate without proof of cause is generally unnecessary and unwise. RBC indices are useful in classifying anemias caused by a production deficit. Their calculation and normal ranges are provided in Table 121-2. MCV is a measure of RBC size; decreases and increases reflect microcytosis and macrocytosis, respectively. MCH incorporates both RBC size and hemoglobin concentration. It is influenced by both and is the least helpful of the indices. The MCHC index is a measure of the concentration of hemoglobin. Low values represent hypochromia, whereas high values are noted only in patients with decreased cell membrane relative to cell volume, such as in the case of spherocytosis. An additional index is the RBC distribution width (RDW), which is a measure of the homogenicity of the RBCs measured. RDW is automatically calculated as the standard deviation of MCV divided by MCV multiplied by 100. A normal RDW is 13.5 ± 1.5%. The RDW is usually elevated in anemias caused by nutritional deficiencies; however, it is not specific for any abnormality. Table 121-2 Calculation of Red Blood Cell Indices and Normal Values Microcytic Anemias.: Hypochromic microcytic anemias are subdivided into deficiencies of the three building blocks of hemoglobin: iron (iron deficiency anemia; Fig. 121-4), globin (thalassemia), and porphyrin (sideroblastic anemia and lead poisoning). Anemia of chronic disease, a secondary iron abnormality, rounds out the differential diagnosis. Not all microcytic anemias are the result of iron deficiency, and routine iron therapy for a patient with a low MCV and MCHC is inappropriate. Iron Deficiency Anemia.: Iron deficiency is a frequent cause of chronic anemia seen in the emergency department. It is the most common anemia in women of childbearing age. In older patients, occult blood loss, especially gastrointestinal, may initially appear as iron deficiency anemia. Because changes in RBC size and hemoglobin content occur only after bone marrow and cytochrome iron stores are depleted, a patient may have early symptoms of iron deficiency (e.g., fatigue) without manifesting changes in RBC structure. The diagnosis is made by laboratory evaluation of the fasting level of serum iron, serum ferritin, and total iron-binding capacity. The laboratory interpretation and pitfalls are outlined in Table 121-3. A concentrated search for occult blood loss is vital. The patient may experience a sense of improvement in as few as 24 hours. Reticulocytosis appears during a 3- or 4-day period in children but may take more than 1 week in adults. The hemoglobin concentration rises on a similar schedule. Explanations for response failures to iron therapy include the following: the patient is noncompliant with the iron supplementation, the blood loss may exceed the replacement, the diagnosis is incorrect, or the diagnosis is partially correct with an additional process complicating the iron deficiency.2 Thalassemia.: Thalassemia is a genetic autosomal defect reflected by the decreased synthesis of globin chains. The hemoglobin molecule is present as two paired globin chains. Each type of hemoglobin is made up of different globins. For example, normal adult hemoglobin (HbA) is made up of two α chains and two β chains (α2β2). HbA2 is α2δ2, and fetal hemoglobin (HbF) is α2γ2. A separate autosomal gene controls each globin chain. Deletions in this globin gene result in an absence or decreased function of the messenger RNA that codes for the creation of that globin. The various globins (α, β, δ, and γ) may be affected by a number of genetic combinations. The decrease in globin production in thalassemia results in decreased hemoglobin synthesis and ineffective erythropoiesis, which is attributable to increased intramarrow hemolysis with destruction of RBCs before they are released. Normal erythropoiesis has a 10 to 20% incidence of ineffective release, with associated intramarrow RBC destruction. This ineffective release of erythropoiesis may double or triple in patients with thalassemia. The cause is believed to be excess chains of the uninhibited globin precipitating in RBCs.3 Although many variations in thalassemia are possible, only three are commonly considered. Homozygous β-chain thalassemia (thalassemia major) occurs predominantly in Mediterranean populations. It represents one of the most common single-gene disorders. The disease is characterized by severe anemia, hepatosplenomegaly, jaundice, abnormal development, and premature death. Patients are transfusion dependent and die as a result of iron deposition in tissues, particularly the myocardium, or infection. Treatment is supportive and consists of transfusion and iron chelation therapy.4,5 Heterozygous β-chain thalassemia (thalassemia minor) is manifested as a mild microcytic hypochromic anemia with target cells (erythrocytes with central hyperchromic bull’s-eye surrounded by pallor) seen on the peripheral smear (Fig. 121-5), an MCV commonly more severely lowered than with iron deficiency anemia, a normal level of serum iron, and an elevated level of HbA2 (α2δ2) on hemoglobin electrophoresis (2-5%). Usually, no treatment is necessary. Therapy consists of blood transfusions, which are based on the clinical severity of the anemia. The goals of transfusion therapy include correction of anemia, suppression of erythropoiesis, and inhibition of increased gastrointestinal iron absorption. Iron chelation therapy is often required to control excess iron stores. Historically, deferoxamine, a subcutaneously administered chelator, was used; however, it has been replaced at many centers with the oral chelator deferasirox (Exjade). Deferasirox is renally cleared; thus dose adjustments may be necessary for chronic renal insufficiency, and it is contraindicated in end-stage renal disease.6 Bone marrow transplantation from HLA-identical donors has resulted in disease-free survival in 60 to 90% of recipients, but its role in thalassemia has yet to be determined. Although much interest centers on permanent correction of genetic deficits in thalassemia, gene therapy does not yet exist.7 Sideroblastic Anemia.: Sideroblastic anemia involves a defect in porphyrin synthesis. The resultant impaired hemoglobin production causes excess iron to be deposited in the mitochondria of the RBC precursor, but some also circulates. The result is increased serum iron and ferritin levels, with transferrin saturation. The defective heme synthesis results in ineffective erythropoiesis, mild to moderate anemia, and a dimorphic peripheral smear with hypochromic microcytes along with normal and macrocytic cells. Anemia of Chronic Disease.: Anemia of chronic disease is common and typically normochromic, normocytic. It is characterized by low serum iron levels, low total iron-binding capacity, and normal or elevated ferritin levels. Bone marrow is normal, but staining reveals an abnormality in the mobilization of iron from reticuloendothelial cells. This anemia can be differentiated from iron deficiency by total iron-binding capacity, serum ferritin level, bone marrow examination, and nonresponsiveness to a trial of iron therapy. Because the hematocrit is seldom less than 25 to 30%, therapy is not usually required. A complete search for occult blood loss is necessary during the evaluation of this diagnosis because iron deficiency may be superimposed. Disseminated cancer, chronic inflammation, uremia, and infection are common causes.8 Macrocytic and Megaloblastic Anemias.: In terms of the potential for a therapeutic response, the most important cause of macrocytosis is megaloblastic anemia. Megaloblastic anemia is the hematologic manifestation of a total-body alteration in DNA synthesis. The defective DNA synthesis is caused by a lack of the coenzyme forms of vitamin B12 and folic acid. The deficiency appears clinically in tissues with rapid cell turnover, including hematopoietic cells and those of mucosal surfaces, particularly in the gastrointestinal tract. This deficiency is characterized hematopoietically by ineffective erythropoiesis and pancytopenia. Vitamin B12 and folate deficiencies have different developmental histories, but the clinical result may be similar. Differentiation of folate and vitamin B12 deficiencies usually depends on measured levels in the laboratory. Folic acid, absorbed in the duodenum and jejunum, is commonly found in green vegetables, cereals, and fruit. It may be destroyed completely by cooking. The body requires approximately 100 µg/day and usually stores 6 to 20 mg. Therefore, a 2- to 4-month supply is available before megaloblastic changes occur. Causes of folate deficiency are listed in Box 121-6. Most patients with folate deficiency have either an inadequate dietary intake, such as alcoholic patients, or increased use, as in pregnancy. Vitamin B12 is found in foods of animal origin only and is not destroyed by cooking. It is absorbed in the ileum after binding to intrinsic factor. This glycoprotein factor is secreted by the parietal cells of the gastric mucosa and allows low levels of B12 to be actively absorbed. The adult requirement is 1 or 2 µg/day, with a body store of 5 mg. Therefore, megaloblastic changes may take up to 4 years to develop after cessation of vitamin B12 uptake. The various causes of vitamin B12 deficiency are listed in Box 121-7. The most common cause is chronic malabsorption. Table 121-4 lists a number of the problems associated with megaloblastic anemia and their underlying pathologic states. A unique feature of vitamin B12 deficiency is its neurologic involvement. Patients may have paresthesias in their hands and feet, decreased proprioception, or decreased vibratory sense. The insidiously developing classic neurologic complex includes loss of proprioception; weakness and spasticity of the lower extremities with altered reflexes; and variable mental changes, such as depression, paranoid ideation, irritability, and forgetfulness. The last two complaints have also been noted with folic acid deficiency. Vitamin B12–deficient patients have some of the lowest hemoglobin levels seen in any disease state. Table 121-4 Clinicopathologic Correlation of Manifestations of Megaloblastic Anemia Macrocytic anemia is suggested when the MCV is greater than 100 fL, but other criteria need to be met for megaloblastosis to be considered the cause of the macrocytic anemia. On the peripheral smear, large oval red cells (macro-ovalocytes) and hypersegmented polymorphonuclear neutrophils are believed to be diagnostic (Fig. 121-6). A bone marrow aspirate may reveal morphologic changes consistent with megaloblastic erythropoiesis. Other potentially useful laboratory tests include vitamin B12 and folate levels, red cell folate, and lactate dehydrogenase (LDH). Laboratory techniques, values, and interpretations are listed in Table 121-5. Once megaloblastic anemia is diagnosed and folate or vitamin B12 deficiency determined, standard diagnostic regimens are followed to determine the precise origin of the deficiency. Because one deficiency may cause gastrointestinal absorption changes that beget other deficiencies, the emergency physician may be forced to initiate therapy before the final diagnosis is made. However, a caution is given to obtain necessary laboratory specimens before this course is pursued. The usual dosage for patients with megaloblastic anemia secondary to folate deficiency is 1 mg of oral folic acid per day. Parenteral administration is generally unnecessary because most cases are due to dietary deficiency. In contrast, malabsorption is the most common cause of vitamin B12 deficiency, and parenteral therapy is initiated at 100 µg/day intramuscularly for the first 7 to 10 days. Thereafter, only monthly 100-µg doses are necessary. The response is often dramatic, with reticulocyte counts rising up 30 to 50% and normalization of RBC, white blood cell (WBC), and platelet counts in 6 to 8 weeks. The use of vitamin B12 or folate supplements in patients with undiagnosed anemia is to be discouraged. The routine injection of vitamin B12 in the elderly has decreased but is still a too common practice.9 Macrocytic anemias unrelated to megaloblastic changes are seen frequently. Liver disease, often associated with alcoholism, is the most common cause.10 Macrocytic target cells may be seen on the peripheral smear in conjunction with this disorder. Hypothyroidism and hemolysis may also be manifested as macrocytic anemia. Screening tests to differentiate between megaloblastic anemia and macrocytic anemia of other causes include a peripheral smear for macro-ovalocytes, hypersegmented polymorphonuclear neutrophils, and the LDH level. Normochromic and Normocytic Anemias.: The origin of normochromic and normocytic anemias secondary to decreased production is not as obvious as that of macrocytic and microcytic anemias because the latter give clues to their origin by alterations in RBC indices. One hematologic parameter that can aid in the diagnosis of normocytic anemia associated with hypoproduction is the corrected reticulocyte count. Reticulocytes reflect RBC production in bone marrow. They are RBCs released from bone marrow every 1 to 3 days and contain residual RNA that can be detected by supravital staining. Reticulocytes have an average MCV of 160 fL and in sufficient numbers can increase the MCV of the total erythrocyte count. The reticulocyte count is expressed as a percentage of the total RBC population and needs to be related (“corrected”) to the RBC count of the patient. Thus the corrected reticulocyte count is equal to the measured percentage of reticulocytes times the patient’s hematocrit (%) divided by 45% (taken as the normal hematocrit). The normal range is 1 to 3%. Aplastic anemia is rare but may have severe manifestations. It is suspected in anemic patients with normal indices, a low reticulocyte count, and a history of exposure to certain drugs or chemicals (Table 121-6). It is related to drug or chemical exposure in 50% of cases. Viral hepatitis, radiation, and pregnancy have been associated with aplastic anemia. Another group of patients is considered to have an “autoimmune” origin. Table 121-6 Aplastic Anemia Caused by Drugs or Chemicals From Silver BJ, Zuckerman KS: Aplastic anemia: Recent advances in pathogenesis and treatment. Med Clin North Am 64:607, 1980. General treatment of aplastic anemia includes removal of suspected marrow toxins from the environment, avoidance of aspirin, oral hygiene, and suppression of menses. Transfusions are given in life-threatening circumstances only. Bone marrow or peripheral blood stem cell transplantation from a histocompatible sibling can cure the bone marrow failure, with survival rates of 78 to 94% reported.10 However, because just 30% of patients have suitably matched sibling donors, only a small number undergo allogeneic transplantation. Immunosuppression with antithymocyte globulin, antilymphocyte globulin, and other cytotoxic chemotherapy is used in the majority of patients who are not stem cell transplantation candidates. Unrelated donors are preferred to avoid sensitization of the patient against the non-HLA antigens that are present in bone marrow from a family donor. The disease has a wide range of severity, and the overall 5-year survival rate is 30 to 40%. Given supportive therapy, up to 80% of patients with severe aplastic anemia still die. Bone marrow transplantation before blood product sensitization has resulted in an 80% 5-year survival rate. This is usually combined with immunosuppressive therapy consisting of antilymphocyte globulin. Difficulty is still encountered in finding the correct immunologic match.11,12 Myelophthisic anemia is bone marrow failure resulting from replacement by an invading tumor, leukemia, lymphoma, or, rarely, granuloma. A more basic defect or inhibitor may complicate the problem because the degree of anemia cannot always be correlated with the extent of bone marrow invasion. Any patient with oncologic disease may be subject to the development of this type of anemia. Useful clues are signs of extramedullary hematopoiesis, such as hepatosplenomegaly and a leukoerythroblastic peripheral smear that demonstrates immature WBCs, nucleated RBCs, and poikilocytosis (teardrop-shaped red cells) (Fig. 121-7). The final diagnosis is made by bone marrow examination. Therapy is directed at the underlying disorder.
Anemia, Polycythemia, and White Blood Cell Disorders
Anemia
Pathophysiology
Diagnostic Findings in Emergent Anemia
Diagnostic Findings in Nonemergent Anemia
Ancillary Evaluation
Differential Diagnosis
Decreased Red Blood Cell Production
INDEX
FORMULA FOR CALCULATION
NORMAL RANGE
Mean corpuscular volume
Hematocrit (%) divided by red blood cell count (106/µL)
81-100 fL
Mean corpuscular hemoglobin
Hemoglobin (g/dL) divided by red blood cell count (106/µL)
26-34 pg
Mean corpuscular hemoglobin concentration
Hemoglobin (g/dL) divided by hematocrit (%)
31-36%
CLINICAL FEATURES
PATHOLOGIC CONDITION
Lemon yellow skin
Combination of pallor with low-grade icterus from ineffective erythropoiesis
Petechiae, mucosal bleeding
Thrombocytopenia
Infection
Leukopenia
Fatigue, dyspnea on exertion, postural hypotension
Anemia
Sore mouth or tongue
Megaloblastosis of mucosal surfaces
Diarrhea and weight loss
Malabsorption from mucosal surface change
Paresthesias and ataxia
Related to myelin abnormality in vitamin B12 deficiency only
< div class='tao-gold-member'>

Full access? Get Clinical Tree


Anemia, Polycythemia, and White Blood Cell Disorders
Only gold members can continue reading. Log In or Register to continue




