(1)
Botulinum Toxin Treatment Program, Yale School of Medicine, New Haven, CT, USA
Abstract
The availability of novel technologies has been crucial in the discovery and description of new pain receptors and modulators expanding our knowledge of the pathophysiology of pain. Data generated from animal models indicate that botulinum toxins can modify and subdue a variety of mechanisms that generate or maintain pain.
The first part of this chapter provides a brief overview of the pathophysiology of pain in light of some of the new developments. The second part provides a focused review of the literature on how botulinum neurotoxins improve and modify animals’ response to pain as well as the therapeutic influence of these toxins on pain receptors, channels, and mediators.
Keywords
Botulinum toxinBotulinum neurotoxinBotulinum toxin ABotulinum toxin BCalcitonin gene-related peptideSubstance PSodium channelsTRPV1 receptorATP receptorDRGRexed layerPeripheral sensitizationCentral sensitizationIntroduction
Over the past 15 years, there has been an outpour of data in the field of pain medicine based on investigations on animals and asymptomatic volunteers. These data have identified new, important pain receptors, promoted our knowledge on pain mediators/modulators, and refined our understanding of pain pathophysiology. Moreover, data emerging from animal studies and human volunteer literature have provided crucial information on how botulinum toxins can influence the pain mechanisms and alleviate pain by altering the function of nerve endings, dorsal root ganglia, and spinal neurons.
In this chapter, the pathophysiology of pain as it pertains to the function of peripheral nerves, DRG, and spinal neurons is discussed first, followed by presentation of the data on botulinum toxin’s effect on pain pathophysiology from the literature on animal studies.
Pathophysiology of Pain at Peripheral and Spinal Levels
Pain-inducing agents (heat, cold, chemical, mechanical) stimulate non-corpuscular, nociceptive free nerve endings and generate action potentials via activity of sodium channels. The generated action potentials, reflecting the nociceptive information, are conveyed to specialized peripheral and central neurons by unmyelinated C and small, myelinated Aδ fibers. C fibers are either peptidergic, using substance P (SP) and calcitonin gene-related peptide as transmitter, or non-peptidergic. The peptidergic fibers end in Rexed lamina I and outer part of Rexed lamina II of the superficial layers of the spinal dorsal horn, whereas non-peptidergic fibers terminate in inner lamina II, Fig. 2.1 (Priestley et al. 2002).
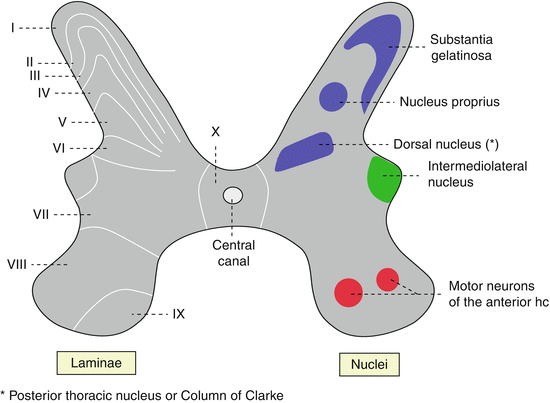
Fig. 2.1
Anatomical location of Rexed lamina and substantia gelatinosa (By User:Polarlys, traced from the German version and translated by Interiot (own work/selbst erstellt) [GFDL (http://www.gnu.org/copyleft/fdl.html), CC-BY-SA-3.0 (http://creativecommons.org/licenses/by-sa/3.0/), or CC-BY-2.5 (http://creativecommons.org/licenses/by/2.5)], via Wikimedia Commons)
In chronic pain, nerve endings exude excess of pain mediators such as glutamate, substance P (SP), and calcitonin gene-related peptides (CGRP) and cause local inflammation. Local inflammation leads to accumulation of inflammatory agents such as bradykinin, cytokines, and prostaglandins. This cascade of events initiates peripheral sensitization of nociceptive nerve endings and the peripheral neuron of dorsal root ganglia (DRG) ultimately lowering the sensitivity threshold of peripheral nerves and neurons to future noxious stimuli. Peripheral sensitization gradually increases the excitability of central sensory neurons and leads to central sensitization of the nociceptive sensory system. The first central nociceptive sensory neuron is located in the two most superficial layers of the posterior horn of spinal gray matter, Rexed areas I and II (Fig. 2.1). Rexed area II or substantia gelatinosa, so named for its paucity of myelinated fibers, modulates pain stimuli in addition to receiving pain signals from the periphery (Melzack and Wall 1965). The excitability of neurons in the superficial Rexed layers is determined by a variety of factors, among them activity of Na+ channels, local excitatory neurotransmitters such as glutamate, and a variety of excitatory and inhibitory receptors, some of them newly discovered (see Fig. 2.2) (Schaible 2007).
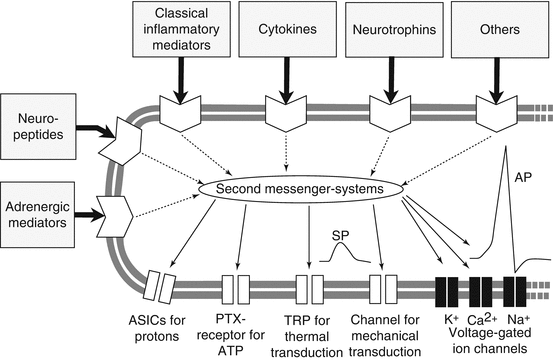
Fig. 2.2
Ion channels and receptors of peripheral nociceptor involved in transmission and modulation of pain signals (From Schaible (2011). Reprinted from Arthritis Research Therapy with permission from John Wiley and Sons, Inc.)
Na+ Channels
Na+ channels, the main generators of action potential, are present diffusely at every peripheral sensory level (receptor, axons, and DRG) and, hence, play a pivotal role in modulating neuropathic and nociceptive pain. A variety of Na+ channels have been described with Na 1.7, Na 1.8, and Na 1.9 most relevant to pain. The Na+ channels are also classified as tetrodotoxin sensitive (TTX-S) with fast activation/inactivation and tetrodotoxin resistant (TTX-R) with slow activation/inactivation. Na 1.7 is a TTX-S channel, whereas Na 1.8 and 1.9 are TTX-R type of sodium channel. Sodium channel mutations are associated with some of the most severe forms of human pain such as seen in erythromelalgia (Fischer and Waxman 2010).
Transient Receptor Potential (TRP) Channels
A major step in understanding the molecular physiology of pain was the discovery of transient receptor potential channels (TRPs) (Caterina et al. 1997). TRPs are expressed specifically in sensory nociceptive neurons. These receptors, made of vanilloid protein (TRPV), are cation-gated calcium channels. Produced by DRG neurons, these protein channels are then transferred by axonal transport peripherally to the nerve endings and centrally to the dorsal horn neurons (Rexed area II, substantia gelatinosa). There are several types of TRP channels designated as TRP1, TRP2, TRP3, TRP4, and TRP8, but TRPV1 plays the dominant role in neuropathic and nociceptive pain. The influx of cations, especially calcium, opens the TRPV1 channel leading to hyperexcitability of the peripheral and central neurons enhancing pain. Heat of over 42°, chemicals such as capsaicin, and low pH of <5.9 directly stimulate and open the TRPV1 channel. A large number of other agents also activate TRPV1 indirectly including inflammatory mediators such as prostaglandin E2, proteases, and nerve growth factor (NGF), with this indirect activation most likely carried through a second messenger (Schaible et al. 2011). The function of TRPV1 channel seems to be different for peripheral (DRG) and central (dorsal horn in SG) neurons. While TRPV1 in DRG neurons receives pain signals from the periphery and conducts the information to SG neurons, activation of TRPV1 in SG neurons releases glutamate locally and promotes central hyperexcitability (Kumamoto et al. 2014).
There is evidence that TRPV1 plays a significant role in production of inflammation-induced hyperalgesia. Inflammatory hyperalgesia is absent in TRPV1 knockout mouse (Davis et al. 2000), and TRPV1 expression is markedly enhanced in neuropathic pain and inflammatory hyperalgesia. Intrathecal injection of TRPV1 antagonist AS1928370 alleviates the neuropathic pain in mouse models (Watabiki et al. 2011). Another TPRV channel, TPRA1, is also upregulated in DRG and dorsal horn neurons by peripheral inflammation and is implicated in cold hyperalgesia caused by inflammation and nerve injury (Obata et al. 2005).
Role of ATP and Purinergic Channels
Purinergic receptors are ligand-gated Ca++ channels that respond to ATP stimulation. The recently discovered P2x3, ATP-responsive receptor channel is specifically expressed in sensory nociceptive neurons. These channels have both chemical and mechanical sensitivity. ATP applied to a blister base causes pain in human and induces pain behavior in animals (Snider and Mc Mahon 1998).
Nerve Growth Factor (NGF)
Nerve growth factor is a major factor in nociception (Snider and Mc Mahon 1998). The development of peripheral nerve endings, C fibers, DRG neurons, and nociceptive sensory spinal neurons is highly dependent on NGF. Specific NGF receptor, tyrosine receptor kinase A (TrkA) is expressed highly in nociceptive neurons. NGF reduces the threshold for heat-generated pain by increasing activity of TRPV1 channels; long-term exposure to NGF increases production of SP and CGRP as well as expression of Na+, P2x3, and TRPV1 channels (Stein et al. 2009). NGF antagonists exert analgesic effects (Lane et al. 2010).
The Role of Spinal Cord GABAergic Neurons in Pain
The excitability of neurons in the superficial laminae of the spinal cord is controlled by GABAergic nerve endings that work upon GABA-A (ionotropic) and GABA-B (metabotropic) receptors attached to the surface of these dorsal horn neurons (Bardoni et al. 2013). The inhibitory input to these neurons comes from inhibitory spinal cord interneurons and from the descending inhibitory fibers. In the state of central sensitization and chronic pain, failure of this GABA system could enhance pain, while enhancing GABA activity could alleviate pain by reducing hyperexcitability of spinal neurons.
Inflammation
As stated earlier in this chapter, with exposure to chemicals and adverse (high or low) temperature and following nerve injury, pain mediators accumulate locally. This accumulation leads to vasodilation and development of inflammation in the damaged tissue. Inflammation, which is associated with lower tissue pH, starts a cascade of events that enhance pain through influencing the function of pain receptors via a variety of mechanisms. Inflammatory cells activate local production of NGF that enhances pain (see above). In acute inflammation, macrophages can directly invade DRG neurons and disturb its function (MacMahon et al. 2006). Lowering of tissue pH by inflammation triggers activity of a specific form of sodium channel (acid-sensing sodium channels) causing hyperexcitability of the neural tissue. Additionally, low pH activates ATP production, which in turn opens the purinergic channels and also activates TRPV1 channels leading to further excitation. The resultant effects are mechanical hyperalgesia and also thermal hyperalgesia due to stimulation of dermal nociceptors.
The Effects of Botulinum Neurotoxins on Pain Receptors, Pain Modulators, Neuroinflammation, DRG, and Spinal Neurons
Over the past 20 years, a large volume of literature has been published on the efficacy of botulinum toxins in modulating pain predominantly based on animal studies. The purpose of this section is not to provide an exhaustive review of the subject, but rather a brief summary of how different nociceptive mechanisms are modified by BoNT resulting in alleviation of human pain. There is currently a significant interest in this subject as reflected in several recent reviews (Aoki 2005; Jabbari 2008; Aoki and Francis 2011; Jabbari and Machado 2011; Guo et al. 2013).
One of the major findings in this line of research was the discovery of significant anti-inflammatory effect for onabotulinumtoxinA (onaA) and subsequent alleviation of inflammatory pain (Cui et al. 2004). A formalin pain model was used as a test model to evaluate the effect of onaA on inflammatory pain and on local accumulation of pain mediators. The authors noted that subcutaneous injection of formalin in rat’s paw produces a biphasic pain response. The first peak of pain develops within 0–5 min of injection and is caused by the direct chemical effect of formalin upon C fibers. The second peak, which occurs within 15–60 min of injection, is a more intense pain induced by tissue inflammation (Wheeler-Aceto et al. 1990). At this stage, a variety of inflammatory agents (neuropeptides, kinins) and pain mediators (glutamate, substance P, CGRP) accumulate at the site of injection. The second peak, unlike the first, is not associated with significant activity of C fibers and is believed to mainly represent central sensitization. Cui et al. (2004) pretreated rats with onaA for 2–12 days in order to observe the time frame of onaA’s effect in formalin-induced pain. Four groups of rats received 3.5, 7, 15, and 30 u/kg of onaA diluted in 0.9 % saline into the hind paw subcutaneously. The control rats received the same volume of 0.9 % saline injected into the hind paw. The rats were then injected with 50 ml of 5 % formalin in the same paw. The immediate lifting/licking (check spelling) behavior was recorded 0–5 min postinjection and, again, at 15–30 min postinjection corresponding to the first and second peaks of formalin-induced pain. The rats were euthanized at the completion of the experiment, and the level of glutamate was measured in the injected tissue.
Related posts:
 Botulinum Toxin Treatment of Myofascial Pain Syndrome and Fibromyalgia
Botulinum Toxin Treatment of Myofascial Pain Syndrome and Fibromyalgia
 Treatment of Dystonic Pain with Botulinum Neurotoxins
Treatment of Dystonic Pain with Botulinum Neurotoxins
 Molecular Structure, Mode of Action, and Immunology of Botulinum Neurotoxins
Molecular Structure, Mode of Action, and Immunology of Botulinum Neurotoxins
 Botulinum Toxin Treatment of Plantar Fasciitis (Plantar Fasciopathy)
Botulinum Toxin Treatment of Plantar Fasciitis (Plantar Fasciopathy)
 Botulinum Toxin Treatment of Chronic Facial Pain: Trigeminal Neuralgia, Temporomandibular Disorders, and Dental-Related Pain
Botulinum Toxin Treatment of Chronic Facial Pain: Trigeminal Neuralgia, Temporomandibular Disorders, and Dental-Related Pain
 Botulinum Neurotoxins and Chronic Low Back Pain
Botulinum Neurotoxins and Chronic Low Back Pain

Full access? Get Clinical Tree


