Chapter 118 Adverse Drug Reactions and Drug-drug Interactions
Adverse drug reactions in the pediatric critical care setting are a potential contributor to the morbidity and mortality of patients.1–5 Although adverse drug reactions in ambulatory pediatric patients may be relatively uncommon, epidemiologic studies in inpatient settings confirm that the problem is just as important for children as it is for adults.3–79 There is substantial evidence that suggests the incidence of adverse drug reactions may be more common than in adults. Many of the therapeutic agents used in the pediatric critical care setting have not been studied and evaluated for safety and efficacy. Intensive care physicians should have a heightened awareness of the incidence of adverse drug reactions so they can assist with recognizing and ultimately preventing adverse drug reactions. Patients in the intensive care unit are predisposed to adverse drug reactions due to the incidence of single and multiple organ failure. Impairment in metabolism (hepatic function) and clearance (renal function) of therapeutic agents increases the incidence of adverse drug reactions. In order to alleviate adverse drug reactions, pediatric intensivists need to be aware of the complications associated with drug therapy in the pediatric critical care patient.
Steps are now being taken to help prevent adverse reactions caused by the lack of knowledge of how a given drug affects children, but remaining issues still cause children to be more susceptible to adverse drug reactions. It is common practice to use weight-based dosing, which adds another layer of complexity to drug therapy in children. For example, many parenteral therapies require dilution of stock solutions, providing an opportunity for errors. Finally, sick infants and children may have reduced ability to survive iatrogenic events if and when they occur. Publications have brought to light the importance of adverse drug events in adults and their impact on the health care system in general.9 Although adverse drug events in ambulatory pediatric patients may be relatively uncommon, epidemiologic studies in inpatient settings confirm that the problem is just as important for children as it is for adults.3–79 Overall, the incidence of adverse events in pediatric inpatients can range from 5% to 21% of hospital admissions,2–5 which is higher than the estimated incidence of approximately 4% in adults.10,11
Defining Adverse Drug Reactions
One of the difficulties in reviewing the literature on adverse drug reactions is how they are defined. There are many different examples of what constitutes an adverse drug reaction. Many professional organizations have tried to standardize exactly how an adverse drug reaction is defined. The World Health Organization (WHO) defines an adverse drug reaction as any response to a drug that is noxious and unintended and that occurs at doses normally used in humans for prophylaxis, diagnosis, or therapy of disease or for modification of physiologic function.12 The Joint Commission on Accreditation of Healthcare Organizations (JCAHO) defines an adverse drug reaction as an undesirable response associated with use of a drug that compromises therapeutic efficacy, enhances toxicity, or both.13
For reporting purposes, the Food and Drug Administration (FDA) categorizes a serious adverse event (events relating to drugs and devices) as one in which “the patient outcome is death, life-threatening (real risk of death), hospitalization (initial or prolonged), disability (significant, persistent, or permanent), congenital anomaly, or required intervention to prevent permanent impairment or damage.”14 The American Society for Health-System Pharmacists (ASHP) defines a significant adverse drug reaction as any unexpected, unintended, undesired, or excessive response to a drug that15:
The ASHP also defines what is not an adverse drug reaction as follows:
The mechanisms by which adverse drug reactions occur can be divided into type A and type B. Type A mechanisms usually are thought of as preventable, predictable, and related to the pharmacologic action of the medication. Knowledge of the effects caused by the medication can predict the likelihood of an adverse drug reaction occurring. Appropriate monitoring of the medication can assist in preventing adverse drug reactions.16–18 Type B mechanisms usually are considered to be unavoidable and not predictable. These reactions normally are uncommon and not related to the pharmacologic action of the medication. Idiosyncratic and hypersensitivity reactions are common type B reactions.16–18
A critical factor in assessing adverse drug reactions is establishing a causal relationship between the suspected drug and the adverse reaction.19 Identification of adverse drug reactions can be arbitrary. Some of the confounding factors are the ambiguous characteristics of the reactions, the fact that patients usually are taking more than one medication at the time of the reaction, and the inability to perform definitive cause and effect tests.19 There have been numerous attempts to try to formalize the process.18–21 Karch and Lasagna19 designed decision tables that can identify a potential reaction, assess the certainty of a link between the drug and the event, and evaluate the underlying cause of the reaction. These provide a framework for systematic evaluation of potential adverse drug reactions and reduce the ambiguity that, at present, characterizes the assessment of adverse drug reactions. Kramer et al.21 devised an operational assessment using an algorithm with six axes to assess the probability of an adverse drug reaction occurrence. A scoring system for each axis and an overall score help delineate the likelihood that an adverse drug reaction has occurred. Similarly, Naranjo et al.18 developed an adverse drug reaction probability scale using 10 questions answered positively (yes), negatively (no), or unknown to determine the likelihood that an adverse drug reaction had occurred. The Naranjo method has been shown to improve the reproducibility of adverse drug reactions assessments.
An FDA algorithm that assesses the characteristics of the event (temporal relationship, dechallenge, rechallenge, and relationship to disease) has been presented.20 The algorithm contains four questions to assess the likelihood that an adverse drug reaction had occurred:
Comparisons of the different methods have been conducted. Michel and Knodel22 compared the Kramer, Naranjo, and FDA methods. The Kramer method was considered to be the standard of practice at the time, and Naranjo and FDA methods both compared favorably. The Naranjo method was shown to be simpler and less time consuming. The lack of a numerical score in the FDA algorithm caused the authors to decide there was a need for more data to recommend its use over Naranjo method, even though the FDA algorithm was less time-consuming than either method.22 Busto et al.23 compared the Kramer and Naranjo methods. The two methods were determined to be equal in terms of reproducibility, but the Kramer method was more complex.
Adverse Drug Reactions by Organ System
Renal
Nephrotoxicity accounts for nearly 7% of all adverse drug reactions.11 Several factors place the renal system at risk for adverse drug reactions. The renal system is responsible for elimination of many drugs and metabolites, of which several are known nephrotoxins. Additionally, the renal vascular system receives approximately 20% to 25% of resting cardiac output. Therefore the kidneys are exposed to high concentrations of drugs and diagnostic agents. Although the renal system is highly vulnerable to nephrotoxicity, there are only a few mechanisms by which nephrotoxins can induce injury. These mechanisms include hemodynamically mediated nephrotoxicity, tubular necrosis, interstitial nephritis, obstructive nephropathy, and vascular toxicity. Many nephrotoxins can injure the kidney through more than one mechanism.
A variety of drugs are associated with acute tubular necrosis, including aminoglycosides, cisplatin, amphotericin B, radiocontrast media, cyclosporine, and intravenous (IV) immunoglobulins.24–26 Acute tubular necrosis is one of the most common renal disorders associated with drug therapy. Minimizing risk factors for nephrotoxicity with these agents is imperative (i.e., administration of a saline load and repletion of volume have proved beneficial in reducing toxicity).27 Table 118-1 provides a more complete list of medications associated with tubular necrosis.
Table 118–1 Drugs Causing Nephrotoxicity
| Tubular Necrosis | Interstitial Nephritis | Hemodynamic-Mediated Renal Failure |
|---|---|---|
NSAID, Nonsteroidal antiinflammatory drug.
Several drugs are implicated in alteration of renal blood flow. Some of the most common include angiotensin-converting enzyme inhibitors (ACEIs) (e.g., enalaprilat), nonsteroidal antiinflammatory drugs (NSAIDs) (e.g., ketorolac), β-adrenergic antagonists (e.g., propranolol), and calcineurin inhibitors (e.g., cyclosporine).7,28–35 ACEIs can induce renal insufficiency in patients suffering from any process that decreases renal blood flow, including bilateral renal artery stenosis or unilateral stenosis with a single kidney.36 The mechanism involves inhibiting the conversion of angiotensin I to angiotensin II, which results in dilation of the efferent arterioles, with resultant decreased glomerular capillary hydrostatic pressure and reduced glomerular filtration. The incidence of ACEI-induced renal failure in patients with renovascular hypertension ranges from 20% to 38%.34 NSAIDs inhibit prostaglandin synthesis, which leads to reduced renal blood flow and reduced glomerular filtration. The incidence of renal insufficiency is most common in patients with chronic renal disease, hypovolemia, sepsis, and use of concomitant nephrotoxic drugs.28,30–33,35 NSAID-induced renal toxicity is reversible if the toxic agent is discontinued immediately.36 Table 118-1 provides a more complete list of drugs associated with hemodynamic renal failure.
Acute interstitial nephritis is another common source of drug-induced nephrotoxicity. It is reported to cause 3% to 8% of all cases of acute renal failure.37 The clinical presentation of acute interstitial nephritis can appear anywhere between 2 and 44 days after initiation of the offending therapy.38,39 Typical clinical symptoms include fever, skin rash, and flank tenderness.38,39 Common laboratory findings include hematuria, sterile pyuria, and eosinophilia.37 Histologic findings of acute interstitial nephritis include interstitial infiltrate of lymphocytes, plasma cells, eosinophils, and neutrophils.37–39 Prompt discontinuation of the offending drug from therapy is recommended, and administration of corticosteroids may improve recovery.40 Table 118-1 lists some medications associated with acute interstitial nephritis.
Renal tubular obstruction is associated with renotubular precipitation of endogenous products, drugs, and their metabolites. Formation of uric acid precipitates after chemotherapy can result in renal obstruction. Hydrating patients prior to chemotherapy, urinary alkalinization, and use of allopurinol or rasburicase can help prevent uric acid precipitation. Rhabdomyolysis can cause intratubular precipitation of myoglobin and lead to acute renal failure. Terbutaline overdose has resulted in rhabdomyolysis-induced renal failure.41 Drugs associated with formation of crystals include acyclovir, sulfonamides, mannitol, pentobarbital, methotrexate, and dextran.42–45
Aminoglycosides are used frequently for treatment of gram-negative infections. All aminoglycosides have been shown to be toxic to the proximal renal tubules.50 Aminoglycoside nephrotoxicity is related to dose, high trough concentrations, and prolonged therapy.50 The drug-induced nephrotoxicity normally manifests as nonoliguric renal failure.47 Several risk factors for developing aminoglycoside nephrotoxicity include the need for intensive care, decreased albumin, poor nutritional status, prolonged therapy, hypovolemia, pneumonia, shock, preexisting liver or kidney disease, and elevated initial steady-state drug concentrations.47,48 In addition, vancomycin, piperacillin, furosemide, amphotericin B, and cephalosporins when administered concomitantly with aminoglycosides are associated with an increased risk for developing nephrotoxicity.47 Predicting nephrotoxicity based on the risk factors stated is an extremely complicated, inexact task.47,49 Typically, aminoglycoside nephrotoxicity is reversible upon discontinuation of the offending agent.50,51
Drug-induced nephrotoxicity is a common serious adverse drug reaction that can lead to morbidity and lengthen hospital stays.52 It is important to recognize potential nephrotoxins before initiating therapy and to evaluate therapeutic options. It is also important to monitor therapy appropriately for signs of toxicity and to modify therapy as needed.
Hepatic
The liver is the most critical organ in drug toxicity due to its major role of metabolism and elimination of foreign substances. This renders it a target for drug toxicity. It is well documented that acetaminophen causes intrinsic hepatotoxicity following acetaminophen overdose. However, a variety of medications cause drug-induced hepatotoxicity. It often is difficult to determine the source of unexplained liver injury, so detailed examination of past and current drug therapies is needed to rule out possible drug-related causes. Most hepatotoxicity associated with drugs is idiosyncratic in nature and has a rate usually less than 1 per 10,000 exposed patients. The pathogenesis of drug-induced liver disease is mediated either through an immune response or direct cell damage.53,54 In many cases, this hepatotoxicity is idiosyncratic, meaning not predictable or dose-related, in contrast to drugs like acetaminophen and isoniazid/iproniazid, for which toxicity is related to dose.53,54
The mechanism of toxicity for drug-induced liver injury generally involves the accumulation of drugs or their metabolites which cause direct cell damage, target mitochondrial function, or trigger an immune response. The initial stress or immune reaction leads to mitochondrial permeability transition, which is either a direct pathway (intrinsic) mediated by cell damage or is triggered by the death receptor amplified pathway. The mitochondrial impairment will determine if the hepatocytes dies by apoptosis or necrosis. Measurements of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), and total bilirubin are important measurements in assessment of drug-induced liver injury. It has been estimated that one tenth of the Hy’s law cases (defined as bilirubin elevated to twice the upper limit of normal [ULN] and transaminase three times ULN) with hyperbilirubinemia or jaundice die or require transplant.55,56 In severe cases of drug-induced hepatic injury, jaundice and hepatic failure may ensue.54 Typically drug-induced hepatic disease occurs expectedly as a result of high-dose therapy of a known hepatotoxin or unexpectedly as a result of an idiosyncratic reaction to a drug not associated with hepatotoxicity. Overall, children are less prone to drug-induced liver disease than are adults.58 However, specific hepatotoxins have been shown to afflict children more than adults. Reye syndrome is a hepatocellular disease that has been associated with aspirin use in children.59 Valproate hepatotoxicity occurs more frequently in children younger than 2 years who have preexisting neurologic or physical defects.60
Drug-induced hepatic disease can occur by a variety of mechanisms, including hepatocellular, cholestatic, and vascular. These injuries are associated with intrinsic or idiosyncratic hepatotoxins. Intrinsic hepatotoxins typically show a dose-related toxic effect. Idiosyncratic hepatic disease is an unpredictable reaction that can occur by immune-mediated hypersensitivity, metabolically or both. Hypersensitivity reactions in the liver can cause cell injury to hepatocytes. The reactions do not correlate with dosage and are difficult to anticipate. This immune-mediated reaction results from antigenic complexes that stimulate T lymphocytes, which can result in hepatic injury. These reactions can be accompanied by fever, skin rash, lymphadenopathy, and eosinophilia.54 The reactions typically resolve upon discontinuation of therapy and resurface when therapy is rechallenged.54 Metabolic idiosyncrasy occurs when a compound that is metabolized to nontoxic metabolites and eliminated in the majority of patients is metabolized to a toxic metabolite in a small number of patients because of genetic differences in metabolism (such as with isoniazid).60 Box 118-1 lists a number of agents that are associated with drug-induced hepatotoxicity.
Cardiovascular
Adverse cardiovascular reactions have been noted to be one of the most common types of adverse drug event in pediatric patients.61 Several medications are associated with adverse cardiovascular effects and the mechanisms are often thought to be related to the agents’ pharmacologic action. These effects are potentiated by four general physiological pathways: electrolyte and fluid imbalances; receptor-mediated sympathomimetic effects; alterations of the electrophysiology of the cardiac tissue; and drug-induced cardiotoxicity.
Medications can affect electrolyte and volume status by direct action on electrolyte receptors or indirectly by affecting prostaglandin release or the renin-angiotensin-aldosterone system. Dramatic changes in fluid and electrolyte status caused by these medications can lead to severe changes in blood pressure, heart rate, or cardiac rhythms. Nonsteroidal antiinflammatory drugs, corticosteroids, diuretics, nesiritide, fenoldopam, and certain immunosuppressant agents, such as cyclosporine and tacrolimus, are all medications that can alter fluid and electrolyte balance.62–67
Pediatric patients in the intensive care unit often require vasopressive medications; however, it is important to be aware that many cardiovascular toxicities have been observed with use of vasopressors and most of these toxicities stem from their receptor-mediated effects (Table 118-2). The α-adrenergic agonism of the sympathomimetics produces palpitations, ectopic heartbeats, sinus tachycardia, and ventricular arrhythmias.68 The direct effect on myocardial tissues may be manifested as electrocardiographic (ECG) changes, such as a reduction in T-wave amplitude, which have been reported during epinephrine infusions in normal individuals. Increased myocardial oxygen demand may precipitate an infarction, especially in individuals with underlying cardiac disease. A dramatic rise in heart rate and peripheral vascular resistance may produce severe hypertension, which could lead to cerebral hemorrhage or hemiplegia. Excessive α-adrenergic stimulation may produce vasoconstriction so severe in the extremities, kidneys, or liver that tissues become ischemic or necrotic. Gangrene of the extremities is reported with high doses of dopamine in patients with underlying occlusive vascular disease. Vasopressin can cause severe vasoconstriction and has been associated with digital loss and splanchic ischemia.69 Extravasation of most sympathomimetic agents may lead to tissue necrosis.70 The sedative dexmedetomidine, though not classified as a vasopressor, has α2-adrenergic agonistic properties and has effects similar to the medication clonidine. When using dexmedetomidine, it is important to monitor patients for hypotension, arrhythmias, and bradycardia.66a
Table 118–2 Adverse Effects of Vasopressor Medications by Adrenergic Receptor Affinity
| Medication | Affected Adrenergic Receptor | Associated Adverse Effect |
|---|---|---|
| Dopamine | ||
| Dobutamine | β1: selective | Increased heart rate, arrhythmias, ↑ myocardial oxygen demand |
| Epinephrine | Vasoconstriction, tachycardia, arrhythmias, tissue ischemia, cerebral hemorrhage, ↑ myocardial oxygen demand | |
| Isoproterenol | β: nonselective (β1 > β2) | Tachycardia, arrhythmias, hypotension, ↑ myocardial oxygen demand |
| Norepinephrine | α and β1 (α > β1) | Potent vasoconstriction, arrhythmias, tissue ischemia Extravasation causes necrosis |
| Phenylephrine | α | Systemic vasoconstriction, hypertension, bradycardia |
| Vasopressin | V1 vascular receptor | Potent vasoconstriction, tissue ischemia |
From Goembiewski JA: Vasopressors used in the crtical care setting, J Peri Anesth Nurs 18:414-416, 2003 Vetter VL, editor: Pediatric cardiology: the requisites in pediatrics, Philadelphia, 2006, Mosby Elsevier.
As noted above, ECG changes can be the side effect of many medications through various mechanisms of action and may lead to dangerous arrhythmias Ventricular arrhythmias in infants and children, unlike adults, are relatively uncommon. The most commonly reported arrhythmias in pediatric patients include conduction disturbances or supraventricular tachyarrhythmias (e.g., atrial tachycardia and junctional tachycardia). A common class of agents whose cardiovascular toxicities stem from its effect on cardiac rhythms are the cardiac glycosides. Digoxin can produce a large array of dysrhythmias; the most common in adults includes unifocal or multifocal ventricular premature complexes and nonparoxysmal atrioventricular junctional tachycardia.71 Sinus bradycardia and a sick sinus syndrome-like effect have been reported as digoxin-induced adverse effects.72 The narrow therapeutic index of this drug class is of special concern in the pediatric population. Another class of medications that requires close ECG monitoring is the antiarrhythmic medications. These medications have mechanisms of action that manipulate cardiac action potential and therefore have the ability to produce dysrhythmias.73,75 The most common types of dysrhythmias associated with the antiarrhythmic medications in all classes are bradyarrhythmias, torsades des pointes, and reentry rhythms.75 An increased incidence of such problems in patients treated for attention deficit and hyperactivity disorder also warrants heightened surveillance for adverse cardiac effects in this population. The central nervous stimulants used to treat this condition can lead to tachyarrhythmias and have been associated with sudden cardiac death.76
Although ventricular arrhythmias are uncommon in pediatric patients, the risk of torsades de pointes is still of concern in this population. Torsades de pointes is a life-threatening subset of arrhythmias and is characterized on electrocardiogram by lengthening of the QT interval and waxing/waning QRS amplitude. Certain medications can increase the risk for this arrhythmia by blocking potassium conductance on the rapid (Ikr) and slow (Iks) receptors within potassium ion channels. This creates a delayed ventricular repolarization and can lead to multiple ventricular reentrant loops. Risk factors for drug-induced torsades de pointes include high doses of associated medications (Box 118-2); underlying structural heart disease; prolonged QT interval at baseline; prolonged QT noted immediately after giving associated medication; and concurrent hypokalemia or hypomagnesemia.78 A notable exception to these risk factors would be the medication quinidine. Torsades de pointes is typically noted at lower medication concentrations and can be observed later in therapy. Most commonly, the antiarrhythmics, tricyclic antidepressants, and fluoroquinolones are general classes of agents that demonstrate the potential to prolong the QT interval.79,80 Another concern when administering these medications is pharmacokinetic drug-drug interactions or pathophysiologic alterations in elimination. Any biochemical mechanism that increases the concentration by inhibition of cytochrome isoenzymes or decreased clearance of the associated medication has the ability to potentiate this type of arrhythmia.81 Recently, several medications have been removed from the market due to fatal arrhythmias secondary to torsades de pointes. The FDA now requires all medications to be screened for their affinities to Ikr/Iks receptors to determine the potential for torsades de pointes. Current practice guidelines recommend avoiding high-risk medications if the patient has a baseline QTc greater than 450 msec and to discontinue or reduce the dose of the medication if the QTc increases to 560 msec or greater after initial medication administration.78
Box 118–2 Drugs Commonly Associated with QT Interval Prolongation
Modified from Crouch MA, Lymon L, Cassano AT: Clinical relevance and management of drug-related QT interval prolongation. Pharmacotherapy 23(7):881-908, 2003.
Miscellaneous
All unmarked medications are considered low risk; however, they have a documented or theoretical association with QT prolongation. For a complete list see www.qtdrugs.com.
Anthracycline antineoplastic agents illustrate a specific pattern of direct toxicity to myocardial tissue. Drugs in this class include doxorubicin, epirubicin, and idarubicin. Acute cardiotoxicity may be manifested as arrhythmias, whereas long-term adverse effects are generally related to the magnitude of cumulative drug exposure, with cardiomyopathy being most common. There may also be the delayed development of cardiotoxicity, as children who have endured anthracycline therapy may exhibit signs of abnormal cardiac function anywhere between 4 and 20 years after exposure.82 These defects include abnormal right ventricular wall motion, impaired myocardial growth and contractility, conduction abnormalities such as QT prolongation, and congestive heart failure.82 Severe anthracycline toxicity cannot be reversed, and such a diagnosis leads to a poor prognosis and high mortality. Heart transplantation may be the only option in such cases.82 Due to the severity of anthracycline cardiac toxicity and the clear association with total cumulative dose received, anthracycline lifetime dosage should be documented for each patient so that cumulative dose thresholds are not exceeded.
Central Nervous System
Prescription medications are the most common cause of iatrogenic seizures.83 Drug-related factors that may contribute to seizures include the intrinsic epileptogenicity of the agent, factors that influence serum levels (dose, schedule, route), and factors that affect central nervous system (CNS) drug levels (lipid solubility, molecular weight, ionization of the drug, protein binding, transport by endogenous systems).84 Patient-related factors may influence the risk for drug-induced seizures, such as preexisting epilepsy, neurologic abnormality, decreased drug elimination capacity (renal, hepatic), and conditions that disrupt the blood-brain barrier.84 There are several medications used within the intensive care unit setting that have been implicated to cause seizures. Due to the complexity of care for many intensive care patients, increasing practitioner awareness of the medications that can induce seizures, as well as their mechanisms, should allow practitioners to recognize and diagnosis patients more effectively (Table 118-3).
Table 118–3 Medications That Can Induce Seizures
| Medication | Potential Mechanism of Action | Comments |
|---|---|---|
| Acyclovir306 | Neurotoxicity | Renal impairment and parenteral administration increases the incidence |
| Antipsychotics307 | Decreases seizure threshold | Increased incidence with high doses and rapid dose adjustments |
| Carbapenems308 | Potentiates seizure activity through the inhibition of GABA receptor | |
| Cephalosporins309 | Inhibition of GABA receptor | Ceftazidime > ceftriaxone > cefuroxime and cefotaxime |
| Cyclosporine310 | Unknown but may involve the release of the potent vasoconstrictor endothelin from endothelial cells which induce microvascular damage and/or changes in permeability of the blood-brain barrier | More common with high doses Increased incidence with hypocholesterolemia and hypomagnesemia |
| Cytarabine311 | Cerebral dysfunction | Increased incidence with intrathecal administration |
| Fluoroquinolones312 | Unknown but may involve inhibition of GABA receptor | History of seizures and use of NSAIDs associated with increased risk of seizures |
| Flumazenil313 | May precipitate benzodiazepine withdrawal; inhibition with seizure medications | Use in caution with patients with underlying seizure disorder |
| Meperidine83,307 | CNS excitation from toxic metabolite (normeperidine); blockade of serotonin | Common reaction with renal impairment and high doses |
| Metronidazole309 | Penetrates the blood-brain barrier | Cumulative high dose and prolonged use |
| Penicillins314 | Irritation of nervous system | Use in caution with infants, rapid IV infusions, and renal impairment |
| Tacrolimus315–317 | Neurotoxicity | May be secondary to local vasoconstriction mediated by endothelin; elevated levels and hepatic impairment may increase the risk of neurotoxicity |
| Theophylline83 | CNS toxicity | serum concentrations >25 µg/mL have been noted to induce seizures |
Phenothiazine tricyclic antidepressants (chlorpromazine, thioridazine) may cause seizures.83 Meperidine may induce seizures because of accumulation of the normeperidine metabolite, especially in patients with renal insufficiency.83 Rapid administration of fentanyl can cause seizures.83 Several antiinfective and immunosuppressive medications, such as the β-lactam antibiotic, imipenem; high-dose metronidazole; isoniazid; cyclosporine; and chlorambucil induce seizures.83 Any of the antiepileptic drugs themselves can be implicated in worsening seizure activity. This usually occurs when higher than normal concentrations are used. Seizures also may be precipitated by medication withdrawal.84 Withdrawal of antiepileptic drugs may cause seizures because of either subtherapeutic levels or too rapid removal of the agent.
Drug-induced headache can be caused by many medications. Headaches can be acute or chronic. They may be caused by use and abuse of the agent.85 Headache may occur during substance withdrawal. Many medications cause acute headaches, including NSAIDs (e.g., indomethacin), vasodilators (e.g., β-blockers, calcium channel blockers, angiotensin-converting enzyme inhibitors), immunomodulators (e.g., cyclosporine, tacrolimus, antithymocyte globulin), cytotoxic agents (intrathecal methotrexate, interferon-α1, interleukin-2), and antimicrobials (e.g., amphotericin-B, tetracycline, sulfonamides).85,86 Chronic headaches may be caused by overuse and abuse of simple analgesics, narcotics, and combination analgesics with barbiturates/sedatives and ergotamine.86
Ototoxicity usually is iatrogenic.77 The main sites of action of ototoxic drugs are the cochlea, vestibulum, and stria vascularis.87 Several medications cause ototoxicity, with salicylates and aminoglycosides the most commonly implicated.87,88 Other agents involved are cisplatin, loop diuretics, erythromycin, and vancomyin.87,88 The effect is usually dose-dependent.77,87,88 High-dose and long-term therapy are common risk factors for aminoglycoside-induced ototoxicity.87,88 For aminoglycosides, streptomycin is mostly vestibulotoxic and neomycin is more cochleotoxic.88 Cisplatin is the most ototoxic of the antineoplastic agents.88 Ethacrynic acid has the greatest potential for causing ototoxicity for the loop diuretics.88 Appropriate dosing and monitoring of agents associated with ototoxicity are important for prevention and early detection.
Hematologic
The most common and serious adverse drug reaction resulting from chemotherapy with cytotoxic agents is bone marrow suppression. The decline in hematopoiesis results from injury to rapidly dividing stem cells and can lead to secondary morbidities. For example, neutropenia predisposes a patient to opportunistic infections and sepsis. Chemotherapy-induced thrombocytopenia may render a patient vulnerable to hemorrhage in the CNS or gastrointestinal (GI) tract. Severe anemia may lead to dizziness, fatigue, hypotension, and myocardial infarction.89 In addition to bone marrow suppression, some antineoplastics may produce thrombotic and hemorrhagic coagulation toxicities. For example, L-asparaginase has been reported to induce changes in the von Willebrand factor multimer in children and thereby promote platelet aggregation.82
Respiratory
Respiratory adverse effects are relatively uncommon but are among the most serious adverse events. Due to the seriousness of some of the drug-induced respiratory adverse events, which could be life-threatening, intensive medical interventions are often required. Numerous agents have been known to cause lung disease. In 1975, only 19 agents were implicated with pulmonary complications.90,91 There are now over 350 agents that have been identified as having the potential to induce respiratory complications.90,91 The number of agents known to cause pulmonary adverse reactions continues to expand each year. Recognition of drug-induced pulmonary disease may be challenging due to the presence of confounding variables often associated with these patients that may result in the underdiagnosis of both acute and chronic lung disease.92 Some of the confounding factors in the diagnosis of drug-induced pulmonary disease include preexisting pulmonary conditions such as bronchopulmonary dysplasia and asthma,93 the use of oxygen,94 and concurrent medications.
Bronchoconstriction is one of the most common drug-induced pulmonary reactions. The two main risk factors for the development of drug-induced bronchospasm include a previous history of reactive airway disease and chronic obstructive lung disease.93,95 There are several classes of medications that are associated with this clinically significant reaction. Analgesics and NSAIDs are two of the most common medications that can precipitate bronchospasm.95 Other agents include antimicrobials (e.g., sulfonamides), cardiovascular agents (e.g., β-adrenergic antagonists and ACEIs), and excipients (e.g., preservatives, coloring agents, antioxidants).95,96 The extensive list of medications suspected of causing or exacerbating bronchospastic reactions demonstrates the need to be aware of drug-induced causes of acute episodes, especially in patients with a history of preexisting lung conditions.
Opiates are commonly prescribed to pediatric intensive care patients. Acute respiratory failure secondary to a noncardiogenic pulmonary has been reported with the use of opiates including methadone, morphine, and codeine.96 Aspirin has also been identified to cause pulmonary toxicity. High concentrations of aspirin can lead to increased vascular permeability causing pulmonary edema. A syndrome of massive fluid retention and pulmonary edema may be seen with interleukin-2 administration. All-trans-retinoic acid may cause acute lung disease secondary to massive total body fluid accumulation.
Chronic pneumonitis/fibrosis is a relatively common syndrome associated with drug-induced pulmonary reactions and is manifested by slowly progressive symptoms of cough and dyspnea.96 Nitrofurantoin is associated with pulmonary fibrosis, most commonly in patients receiving chronic, high-dose therapy and/or patients with renal insuffiency.97 A delayed presentation (months to years after discontinuation of or the start of therapy) is the most common form of fibrosis seen with amiodarone,98 but it also may cause an acute fibrosis manifesting within the first few weeks of therapy.97,98 Many cytotoxic agents have been known to cause pulmonary fibrosis including bleomycin, carmustine (BCNU), mitomycin, mitomycin/vinca alkaloid combination therapy, and cyclophosphamide. Risk factors for this reaction to cytotoxic agents are cumulative dose, age at treatment, and radiation therapy.
Endocrine and Metabolic
Due to the complexity of the biochemical processes that influence the endocrine and metabolic balance of the body, several medications have the potential to create alterations in neuroendocrine hormonal production, binding, transport, and signaling. Additionally medications may create changes in hormonal counter-regulatory efforts. The most common types of medication-induced endocrine disorders are modifications in carbohydrate metabolism, electrolyte and calcium abnormalities, drug-induced thyroid changes, and alterations in acid-base status.99 Variations in carbohydrate metabolism are commonly seen with many medications and are clinically manifested as alterations in blood glucose. Table 118-4 lists the multitude of medications associated with hypoglycemia or hyperglycemia subdivided by neuroendocrine effect.99–109
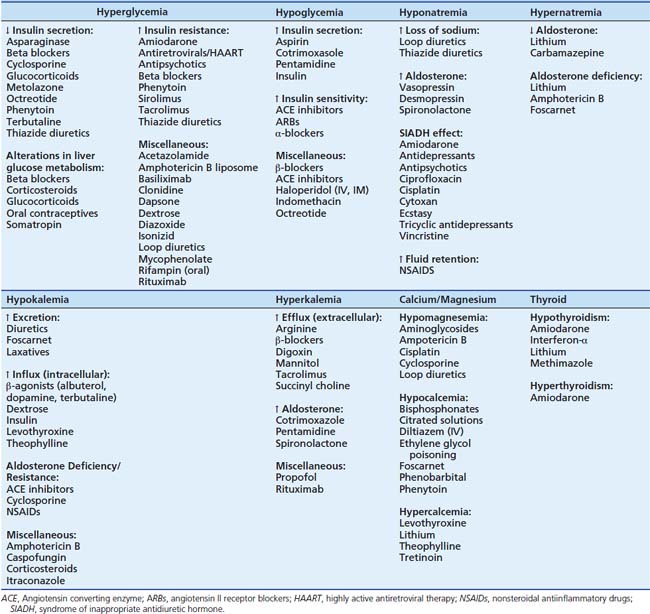
Hypoglycemia occurs most commonly with overtreatment with insulin or diabetic medications, while hyperglycemia results from medications that either increase glucose production or decrease the effects of exogenous insulin.99,110,111 Decreased insulin secretion, changes in liver glucose metabolism and production, and increased insulin resistance are the most common mechanism of drug-induced hyperglycemia.99 Glucocorticoid usage is commonly associated with hyperglycemia and glycosuria.112 Corticosteroids increase blood glucose via hepatic gluconeogenesis and by increasing peripheral insulin resistance.113 This adverse drug reaction appears to be dose-dependent and usually is reversible upon discontinuation of therapy.113 Patients on high-dose, long-term glucocorticoid therapy are at significant risk for suppression of the hypothalamic-pituitary-adrenal axis.114,115 Symptoms of adrenal insufficiency include arthralgias, dizziness, hypotension, nausea, and weakness.114
Drug-induced electrolyte disturbances can occur with a variety of pharmaceutical agents. Several medications are associated with initial or worsening hypokalemia, hyperkalemia, hyponatremia, hypernatremia, and hypomagnesemia (see Table 118-4).99,116–131 Hyponatremia and hypernatremia are often secondary to depletional or dilutional effects of medications or drug-induced changes in antidiuretic hormone.99,118 Hypokalemia is directly affected by medications that increase potassium excretion or that increase Na-K-ATPase activity, causing potassium influx into the cells. Drug-induced hyperkalemia can develop when medications lead to decreased Na-K-ATPase activity, aldosterone deficiencies, and resistance to aldosterone.99,116 Medications that affect the renal tubules of the kidneys, such as amphotericin B and cyclosporine, have been associated with severe hypomagnesemia.120 Antiepileptic agents, citrated solutions, foscarnet, and intravenous diltiazem have all been associated with critical hypocalcemia due to effects on either vitamin D metabolism or calcium reuptake mechanisms.121–127 Electrolyte disorders represent a common adverse drug reaction and can greatly complicate therapy. Accordingly, frequent monitoring of electrolytes is warranted for patients on multiple drug therapy.
Electrolyte abnormalities are noted with the use of amiodarone; however, it is most commonly known for its adverse effects on the thyroid gland. Due to the large amount of iodine in amiodarone (3 mg of iodine/100 mg of drug), it has a counterregulatory effect on the production of thyroid hormones.99 In patients in more developed countries, where iodine-deficiency is rare, use of amiodarone is associated with hypothyroidism; however, in countries where iodine-deficiency is more common; amiodarone can cause thyrotoxicosis.129,130 It is important to obtain baseline thyroid studies prior to starting therapy and to continually monitor them throughout treatment.
Although uncommon, prolonged use of nitroprusside can result in methemoglobinemia and cyanide or thiocyanate toxicity.132,133 Thiocyanate or cyanide toxicity is uncommon but can occur in patients suffering from kidney or hepatic failure or in patients receiving high-dose or prolonged infusions of nitroprusside.134 An early indicator of thiocyanate or cyanide toxicity is metabolic acidosis.135 Other medications associated with anion gap metabolic acidosis include chloramphenicol, epinephrine, norepinephrine, papaverine, and salicylates.136,137
Several life-threatening adverse drug reactions with propofol use in both the pediatric and critical care setting have been reported. In 2001, the FDA communicated a warning against off-label use of propofol for sedation in pediatric intensive care.138,139 The FDA concern came from reviewing data of a randomized, controlled clinical trial evaluating the safety and efficacy of propofol versus standard sedation regimens in pediatric intensive care patients. Approximately 10% of patients treated with propofol died compared with 4% of children receiving standard treatment. Although large case series have reported the safe use of propofol without adverse effects,140 given the risks of mortality with propofol infusion syndrome, prolonged propofol infusions are not recommended in the pediatric ICU setting.
Dermatologic
Erythema multiforme major, Stevens-Johnson syndrome, and toxic epidermal necrolysis are severe cutaneous reactions. They are characterized by the triad of symptoms of mucous membrane erosions, target lesions, and epidermal necrosis with skin detachment.141 Drugs are frequently cited as the cause of these reactions. The more severe the reaction, the more likely it is drug-induced.141 Numerous medications are implicated as the cause of these reactions, such as antiepileptic agents, antibiotics (e.g., sulfonamides), allopurinol, and NSAIDs (e.g., piroxicam).141,142 For most agents, the pathogenesis is unknown. However, with sulfonamides and antiepileptics, the cause can be linked to a toxic metabolite that, in predisposed patients with a genetic defect, can bind covalently to proteins and elicit an immune response that causes a cutaneous reaction.141
Hypersensitivity syndrome reaction is a multisystem idiosyncratic reaction that manifests as fever, rash, and symptomatic or asymptomatic involvement of internal organ systems (liver, kidney, lungs, spleen, muscles, pancreas, lymph nodes, and blood).142,143 Reactions usually occur with first exposure to medication, with initial symptoms occurring 1 to 6 weeks after exposure.141 Aromatic antiepileptic agents (carbamazepine, phenobarbital, phenytoin, primidone) form a toxic metabolite that causes a secondary immune or hypersensitivity reaction.143,144 Lamotrigine, valproic acid, and ethosuximide are other antiepileptics that may cause reactions. Sulfa drugs (dapsone, sulfasalazine, sulfonamide), allopurinol, and diltiazem also are implicated in hypersensitivity reactions.143
Drug rashes are the cutaneous manifestation of drug hypersensitivity. “Red neck” or “red man” syndrome, which is associated with the rapid infusion of vancomycin, is a histamine reaction and not a true allergic reaction. Prolongation of the administration prevents this reaction. β-Lactams and sulfamethoxazole are other common agents that cause rash.97
β-lactam antibiotics (most commonly) and sulfamethoxazole are medications known to cause anaphylaxis. The signs and symptoms of this immunoglobulin E–mediated allergic reaction are pruritus, urticaria, cutaneous flushing, hives, angioedema, bronchospasm, nausea, vomiting, diarrhea, nasal congestion, rhinorrhea, laryngeal edema, and hypotension. Several medications are associated with a drug-induced lupuslike reaction. Procainamide, hydralazine, isoniazid, nitrofurantoin, and griseofulvin are some of the agents involved.97,141 Determination of the reaction is based on symptom resolution with drug discontinuation, absence of idiopathic lupus, development of antibodies, and one clinical symptom of lupus.141
Drug-drug Interactions
The pediatric intensive care physician is confronted daily with potentially hazardous drug-drug interactions in the critical care setting. A potential drug-drug interaction can be defined as “the possibility that one drug may alter the intensity of pharmacological effects of another drug given concurrently. The net result may be enhanced or diminished effects of one or both of the drugs or the appearance of a new effect that is not seen with either drug alone.”145 The ability to recognize every potential drug interaction in the pediatric critical care setting is nearly impossible. Given the extent of polypharmacy that occurs, the potential risk is significant. Of the thousands of documented drug-drug interactions, only a small fraction are clinically significant.146 The ability to differentiate between clinically significant and insignificant interactions requires an understanding of their mechanisms. In most cases, drug-drug interactions are predictable and preventable with appropriate dosage modifications or avoidance of combinations.
Drug-drug interactions can occur by three different mechanisms of action: pharmacokinetic, pharmacodynamic, and pharmaceutical. Pharmacokinetic drug-drug interactions can be defined as “interactions which affect a target drug through alterations in their absorption, distribution, metabolism, or excretion; the result may be an increase or decrease in the concentration of drug at the site of action.”145 Pharmacodynamic interactions are defined as interactions at a common receptor site or that have additive or inhibitory effects as a result of actions at different sites.145 Pharmaceutical interactions or incompatibilities “occur when drugs interact in vitro so that one or both are inactivated.”147
Pharmacokinetic Drug-drug Interactions
Interactions Affecting Drug Absorption (Enteral Absorption)
Several factors determine the rate and extent of oral absorption of drug products. Drug-drug interactions affecting enteral absorption occur through several mechanisms with a common end result: alteration of availability of the drug at its primary site of absorption. The most common mechanisms include adsorption or complexation of the target drug by other drugs or by food, alterations in the ionization of the drug through pH changes, and perturbation of normal GI function (e.g., motility, bacterial colonization, mesenteric blood flow). In the critical care setting, adsorption and complex formation are the most likely causes of decreased enteral drug absorption. Commonly prescribed drugs (e.g., sucralfate and kaolin pectin) are implicated in causing decreased absorption of other drugs by adsorbing target drugs and rendering them unavailable for absorption across the GI barrier.147a,148,149 Food, especially enteral feeding products, is capable of causing adsorption of drugs. Considerable controversy surrounds the potential for this interaction in the case of phenytoin; the clinical importance of this interaction is still debatable.150 Although less commonly prescribed in the pediatric setting, tetracyclines and quinolone antibiotics have long been known to cause drug complexes with metallic cations such as iron and calcium, resulting in a decreased effect of both the supplement and the antibiotic.151,152
Because only un-ionized drug is available for absorption through the intestinal (or gastric) mucosa, the pH at the site of GI absorption must be considered. In theory, absorption of weakly basic drugs such as penicillins or sulfonamides would be enhanced when given in the presence of concomitant H2-antagonists or proton pump inhibitors for GI prophylaxis. However, most clinical investigations show this interaction is insignificant.153–155 Logically, weakly acidic drugs such as phenobarbital would be expected to have reduced enteral absorption under the same circumstances; however, this has not been confirmed clinically.
Alterations in the structure or function of the GI tract are not often the cause for drug-drug interactions but nonetheless are important to consider. Drugs that affect gastric emptying or intestinal transit time can reduce or prolong the residence time of a drug at its site of absorption, potentially altering its systemic availability. Alterations in the normal bacterial flora of the gut can affect the absorption of drugs that are metabolized by these bacteria to active (or inactive) forms. Digoxin is the most common example of a drug whose absorption can be affected through this mechanism.156–158 Alterations in mesenteric blood flow potentially could impact the absorption of any drug administered enterally, but this has never been shown clinically. Although controversial, the ability of indomethacin to affect mesenteric blood flow has been cited as a theoretical reason to withhold feeds and enteral drugs in children being treated for patent ductus arteriosus.159,160






Full access? Get Clinical Tree


