There are a myriad of causes that can lead to acute weakness in a patient in the intensive care unit. These include diseases affecting the central nervous system (CNS), the peripheral nervous system (PNS), the neuromuscular junction (NMJ), and the muscles (myopathic disorders). Other etiologic causes include effects of drugs/medication and injuries. Careful consideration hereof is crucial to reach a diagnosis with the aid of a full laboratory assessment along with specific testing such as lumbar puncture, electrophysiological studies (EMG/ENG), chest computed tomography (CT), and the like as warranted.
I. CNS CAUSES
A. Stroke
Patients being treated in a (nonneurological) intensive care unit can present with an acute muscle weakness caused by an ischemic or hemorrhagic stroke. Signs of lateralization and other focal neurological deficits ought to be recognized. Imaging of the brain with CT and/or magnetic resonance imaging (MRI) should be performed as soon as possible to assess the situation (hemorrhage, acute ischemic stroke), and to consider specific treatment (e.g., external ventricular drain, intravenous tPA, mechanical recanalization) when warranted.
B. Infection
Infections (meningitis or meningoencephalitis) and abscesses of the CNS are further causes that could lead to an acute muscle weakness in an intensive care unit (ICU) patient, which could typically go along with high fever, confusion, and focal defects like seizures. Careful evaluation of the serum lab work, cerebrospinal fluid (CSF) studies, and brain imaging is required to reach a diagnosis. Treatment will be goal directed (antibiotics, antiviral meds, surgical treatment for abscesses).
C. Central Pontine Myelinolysis
A progressive tetraparesis along with a decreased level of consciousness and brainstem dysfunctions (dysphagia, complexe III nerve palsy, weakness of respiratory muscles) should prompt the clinician to think of too rapid over- or undercorrection of serum sodium levels leading to central pontine myelinolysis. This osmotic demyelating disease occurs mainly in the pons and is supposed to happen because of dehydration of brain cells in the setting of a too rapid shift of water from the tissue; the exact mechanism is unclear. Extrapontine myelinolysis would include locations such as the cerebellum, basal ganglia, corpus callosum, and internal capsule. MRI would confirm the diagnosis. There is no specific treatment.
II. PNS CAUSES
A variety of conditions impacting the peripheral nervous system can lead to weakness, with the impact ranging from mild and isolated, to broad and severe. Patients with fractures should be screened for any associated nerve impairment. Compression injuries to peripheral nerves may result from suboptimal patient positioning during surgery or periods of prolonged bedrest. Many acquired polyneuropathies such as those resulting from vascular disease, diabetes, or alcohol use occur over time and thus do not typically present as an acute loss of strength. However, it is important to consider such conditions in the weak ICU patient who cannot provide a functional history. Infections, toxins, endocrine disorders, and vitamin B12 deficiency can also lead to polyneuropathy.
A. Guillain-Barré Syndrome. Guillain-Barré syndrome (GBS) describes a syndrome of acute, immune-mediated, inflammatory polyneuropathies that consist of several different forms. GBS is typically preceded by an infection or illness and is characterized by widespread, patchy areas of peripheral nerve demyelination that translates into weakness and paralysis. Acute inflammatory demyelinating polyneuropathy (AIDP) is interchangeably used with GBS. Variant forms of GBS including Miller Fisher syndrome (MFS), acute motor axonal neuropathy, and acute sensorimotor axonal neuropathy are less frequently seen in the US. Clinical presentation includes migrating, symmetrical paralysis, typically starting in the legs with absent or diminished deep tendon reflexes. Patients with MFS present with ophthalmoplegia, ataxia, and areflexia. Paresthesias and neuropathic pain are common. Symptoms usually progress over a 2-to-3-week period before reaching a nadir, followed by subsequent improvement. Degree of weakness can vary from mild to severe with complete paralysis. Approximately 25% to 30% of patients will have sufficient involvement of the respiratory muscles to require mechanical ventilation. Autonomic dysfunction including dysrhythmias, hypotension, and hypertension is common and may be fatal; thus, close monitoring is warranted.
1. Diagnosis is based on clinical exam and confirmed with analysis of CSF and electrophysiology studies. Treatment in the acute phase includes supportive care with particular attention to the respiratory and cardiovascular systems. Response to intravenous vasoactive drugs is often exaggerated and thus should be used with caution. Rehabilitation in the acute phase includes a focus on preventing secondary complications and initiating gentle active exercises, titrating to patient response. More intense rehabilitation is often indicated after the acute phase to restore function. Specific therapies to treat GBS include plasma exchange and IV immunoglobulin (IVIG); treatment with either plasma exchange or IVIG is associated with improved recovery and expedited time to independent ambulation. Remyelination and functional recovery occur over a period of weeks to months. Approximately 80% of patients return to independent ambulation by 6 months; 5% to 10% of patients have delayed and/or incomplete recovery. Relapses occur in approximately 7% of patients and are generally treated with the initial regimen. Deterioration after initial improvement and stabilization, or prolongation of symptoms beyond 8 weeks may indicate the presence of chronic inflammatory demyelinating polyneuropathy (CIDP).
B. Critical Illness Polyneuropathy
Critical illness polyneuropathy (CIP) is a sensorimotor neuropathy characterized by distal axonal degeneration. CIP frequently coexists with critical illness myopathy (see later in chapter). Clinically, these two conditions can be difficult to distinguish, and it is not always necessary to do so. The alternative term “ICU-acquired weakness” (ICU-AW) is used extensively in the literature to describe more broadly weakness detected on clinical exam that develops as a function of critical illness, including both CIP and CIM. ICU-AW is common in critical illness, affecting 25% to 85% of patients, with increasing incidence in those with sepsis and multiorgan failure. The specific etiology of CIP is multifactorial, with inflammation, impaired perfusion, and altered permeability all potentially contributing. Typically CIP becomes apparent as the patient regains arousal and is noted to have profound weakness with inability to wean from the ventilator. It is characterized by flaccid, usually symmetrical weakness and diminished sensation with diminished or absent reflexes. Lower extremities may be more affected than upper extremities, and distal muscle groups more than proximal. Facial muscles are often spared; therefore, it is important to include assessment of facial movements when determining command following. Muscle atrophy will be present.
1. Formal diagnosis of CIP requires electrophysiology testing. Nerve conduction studies (NCS) will show evidence of a sensorimotor axonal neuropathy including decreased amplitude of compound motor and sensory action potentials, with absence of conduction block. Needle electromyography (EMG) will demonstrate resting fibrillation potentials. Bedside manual muscle testing procedures in cooperative patients will often provide adequate support for the diagnosis of “ICUAW” once other causes are ruled out (see Table 31.1). Electrophysiology studies should be considered in cases where a patient cannot be accurately examined at the bedside, and/or where weakness does not show improvement with time.
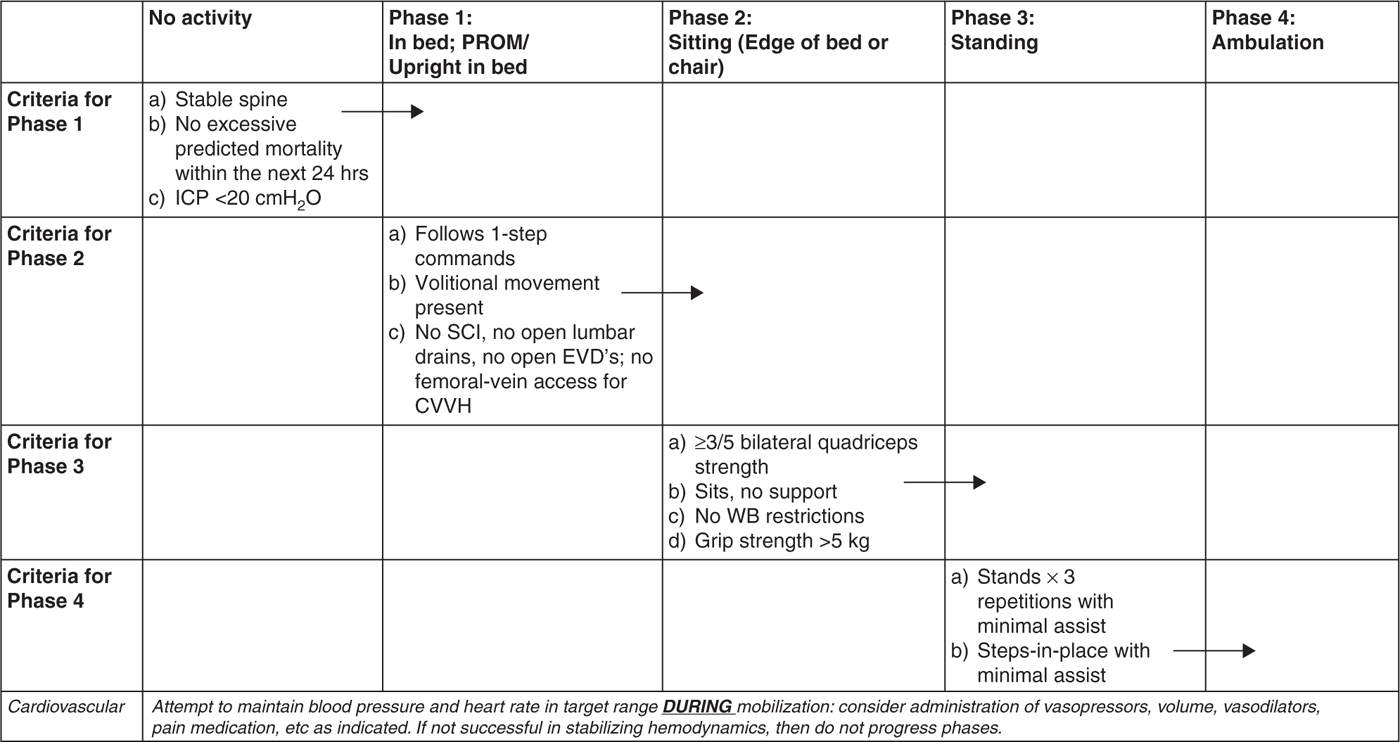
2. Strategies to prevent or minimize the development of ICU-acquired weakness should be incorporated into clinical practice as feasible. While robust, cause-effect evidence is limited, associations have been found among several critical care variables and ICU-AW and CIP. In particular, immobility contributes directly to muscle atrophy, and thus it is key to integrate exercise and mobilization into patient care as early as clinically appropriate. Early mobility pathways or algorithms can be helpful in facilitating safe, targeted, incremental activity in critically ill patients (see Fig. 31.1). Regular screening for participation in spontaneous awakening trials is encouraged in order to minimize oversedation, which in turn perpetuates immobilization. Similarly, when clinically acceptable, it is important to enable spontaneous breathing to facilitate diaphragm activation in order to minimize diaphragmatic atrophy.
a. Prevention efforts may also include aggressive management of sepsis to minimize systemic inflammation and oxidative stress, as these conditions appear linked to ICU-AW and CIP. Consider early nutrition when appropriate to help mediate muscle catabolism. There is some evidence to show that intensive insulin therapy reduces the incidence of CIP; however, because other potential risks exist with this practice, it cannot be broadly recommended. The association between use of neuromuscular blocking agents (NMBA) and development of ICU-AW is not clear-cut. While NMBA have an apparent role in the management of select conditions (e.g., severe ARDS), it is probably wise to avoid prolonged use when possible.
3. Once present, treatment of ICU-AW and CIP ought to include the aforementioned considerations, as well as good supportive care and rehabilitation. Prognosis for recovery with CIP is variable and can be prolonged. CIP is associated with increased length of stay, time on the ventilator, mortality, and reduced functional outcomes. While most patients gradually recover over weeks to months, evidence suggests that a sizable portion of patients are not fully recovered at 1 year, particularly those with severe involvement.
| Manual Muscle Testing Screen for ICU-Acquired Weakness in Cooperative Patients | |
| Right | Left |
Abduction of the shoulder Extension of the wrist Flexion of the hip Extension of the knee Dorsal flexion of the foot |
Sum Score (max 60): Test the six muscle groups listed above, bilaterally in sufficiently alert and attentive patients. Grade each muscle on score of 0–5. All grades can be combined for a “sum-score” out of 60.
Sum-scores ≤48 may be associated with ICU-acquired weakness.
MRC strength grading scale: 0, no muscular contraction; 1, trace or flicker of contraction; 2, active movement with gravity eliminated; 3, active movement against gravity; 4, active movement against gravity and some resistance; 5, active movement against gravity and full resistance.
III. NEUROMUSCULAR JUNCTION DISORDERS
There are different etiologies for neuromuscular junction disorders (NMJD) discussed in the literature whereby myasthenia gravis (MG), belonging to the autoimmune category, is the most common form. Other causes are congenital or toxic (e.g., botulism).
A. Myasthenia Gravis
MG is a disease that interferes with the transmission of acetylcholine at the neuromuscular junction, leading to proximal muscle weakness and fatigue. In the majority of cases, it is caused through the binding of circulating autoantibodies to postsynaptic nicotinic ACh receptors. This in turn prevents acetylcholine, the neurotransmitter that is responsible for muscle contraction at the motor end plate, from connecting to its receptor. There is a generalized, an ocular, and a paraneoplastic variant of MG. The above-mentioned autoantibodies can be found in about 80% of those with the generalized form of MG. In about 10% of the MG patients, a thymoma can be detected, which goes along with anti-titin-antibodies.
1. The lead symptom of MG is general fatigue associated with a progressive proximal muscle weakness, especially upon activity and improving with rest. There is a typical progression during the course of the day, with a peak weakness during the evening hours. Facial, oropharyngeal, ocular, and neck muscles are as susceptible as skeletal muscles. Ocular involvement with diplopia and ptosis is frequently the initial sign. Further symptoms include dysarthria and dysphagia with severe cases affecting the respiratory muscles as well. Myasthenic crisis is a life-threatening condition with respiratory failure and aspiration that develops usually over days, rarely acutely. It is caused by infections, errors in intake of medication, and insufficient immunosuppression. Intensive care support and plasma exchange or IVIG are vital in these cases. Despite these measures, the mortality can be still as high as 5%.
2. A cholinergic crisis can present clinically in a similar fashion to the myasthenic crisis with flaccid paralysis; however, the underlying pathophysiology, and thus the therapy, is very different. Treatment with excess doses of cholinesterase inhibitors can lead to a cholinergic crisis by nonresponsiveness of ACh-receptors to abundant acetylcholine. Applying edrophonium (an ACh-esterase inhibitor) can distinguish both forms of crises by worsening the cholinergic crisis and by improving the symptoms of a myasthenic crisis. There is no specific treatment for cholinergic crisis other than discontinuing the responsible agents and applying supportive measures like intubation and mechanical ventilation. Atropine, a blocking agent at the muscarinergic ACh receptor, has only limited impact on the muscle weakness component, which is triggered through nicotinergic acetylcholine receptors. Several medications can exacerbate symptoms of MG (Table 31.2).






Full access? Get Clinical Tree


