Type of operation
Incidence of chronic post-surgical pain
Number of operations performed in USA in 2006 (DeFrances et al. 2008)
Coronary artery bypass graft
444,000
Hysterectomy
4.7–31.9% (Brandsborg et al. 2008)
560,000
Mastectomy
25–60% (Gartner et al. 2009)
60,000
Thoracotomy
21–61% (Gottschalk and Ochroch 2008)
699,000
Amputation (lower limb)
60–80% (Nikolajsen and Jensen 2001)
65,000
Cesarean section
6–18% (Vermelis et al. 2010)
1,269,000
Cholecystectomy (open + lap)
721,000
For reasons discussed above, establishing an appropriately inclusive definition has been challenging. However, many studies reporting on the epidemiology of certain CPSP syndromes failed to utilize any type of definition. When definitions have been used, they have often been inconsistent (Poobalan et al. 2003 #780).
Epidemiological studies of CPSP have also lacked uniformity in many ways other than definition as well. Clinical variables including the duration of follow-up, surgical technique, patient population, and what, if any, preventative therapeutic measures were undertaken have also differed from study to study. Such discrepancies likely contribute to the wide range of reported incident values (Bruce et al. 2003, 2004).
Further affecting the quality of these epidemiological studies is the fact that most are retrospective. Therefore, they rely on a patient’s ability to accurately remember the amount of pre-surgical, acute post-surgical, and CPSP experienced. Such patient recall has been demonstrated to be unreliable (Tasmuth et al. 1996).
Another interesting phenomenon is that those studies in which CPSP is the primary focus tend to report a higher incidence of post-surgical pain. A striking example of this occurrence involves the reported incidence of phantom limb pain following amputation. Even though the currently accepted rate of chronic phantom limb pain is thought to range between 60 and 80%, some early literature reported incidence rates as low as 2%. This discrepancy seems most likely a result of early studies that based their incidence rates on patients’ requests for pain treatment following surgery, a practice that significantly underestimates the true frequency of CPSP (Nikolajsen and Jensen 2001). It is has been historically demonstrated that patients are often unwilling to complain of phantom limb pain before being prompted due to fear of being labeled as mentally unstable (Sherman and Sherman 1983). While the rate of underreporting may vary depending on the type of post-surgical pain in question, it still begs the question of whether an unrecognized, and possibly undertreated, population of CPSP patients exists.
To date, epidemiological studies have been procedure specific, and thus the overall incidence of CPSP is not known. However, to illustrate the possible overall societal impact of CPSP, it is helpful to consider the estimated number of annual surgeries. According to the CDC National Center for Health Statistics (NCHS), in 2006, there were an estimated 46 million surgical procedures performed in the United States alone. Taking into consideration only the seven procedure types listed in Table 19.1 and applying the lowest incidence rate for each, suggests that as many as 460,000 new patients can potentially experience CPSP each year. If one accepts the highest incidence rate for each procedure, the number of new patients suffering from CPSP each year may be as high as 1.5 million.
Pathophysiology
Pain is an evolved phenomenon comprised of both sensory-discriminative and affective-motivational components that serves the role of protecting an organism from harm. As such, it changes to adapt to the various circumstances and environments encountered. These changes can be either in the form of amplification or attenuation of sensory input, and when operating appropriately, function to promote and enhance survival. The process by which the nervous system alters how it responds to both external and internal stimuli is termed neuronal plasticity. A commonly cited example of extreme attenuation is that of soldiers who continued to function in battle despite sustaining severe injuries, including loss of limb, and later reported that at the time they felt little or no pain (Beecher 1946 #730, 1959 #733). More often recognized is the ability of the nervous system to increase its sensitivity following an injury so that even non-noxious stimuli begin to cause pain, a process termed sensitization. Such a response is appropriate to prevent further injury during the time of healing. Typically, once an injury heals, the sensitivity of the nervous system returns to baseline and pain resolves. However, in some cases the nervous system fails to reset and instead pain continues to be experienced, even long after tissue healing has finished. When this occurs, pain itself becomes the disease.
The mechanisms responsible for neuronal plasticity are multiple, and all levels of the pain signaling pathways are involved. The degree to which sensitization occurs depends on factors such as the intensity and duration of the nociceptive input, degree of inflammatory response from the tissue injury, and magnitude of any direct peripheral nerve injury that may have occurred. There is also growing evidence that genetic, environmental and developmental patterns also play a role in predisposing certain individuals to chronic pain (Papp and Sobel 2009 #777; Al-Chaer and Weaver 2009 #778). The myriad of neuroplastic changes that occur can be broadly categorized into three types based on the mechanism of action. Activation-dependent plasticity is attributed primarily to conformational receptor changes and/or a temporary loss of inhibition and is typically short-lasting. Modulation occurs via post-translational changes to receptor and ion channels and produces longer lasting but reversible increases in the sensitivity of the nervous system. Modification is an increase in the sensitivity of the nervous system that results from transcriptional changes in gene expression. The effects of modification tend to be more permanent (Woolf and Salter 2000).
CPSP is a syndrome based entirely on the fact that sometimes acute pain becomes chronic. Therefore, it is an ideal pain state for questioning how the transition from acute to chronic pain occurs. In fact, many investigators who use animal models to study chronic pain have incorporated surgical procedures into their methodologies for this very reason. This section attempts to provide an overview of neuroplastic changes that contribute to the development of chronic pain. While this chapter focuses on CPSP, the pathophysiology discussed below is applicable to the generalized field of chronic pain.
Basic concepts of pain signaling: Primary afferent fibers can broadly be classified based on their conduction velocity (which is dependent on size and the presence or absence of myelination) and the stimuli by which they are activated. A-beta (Aβ) fibers are large, myelinated fibers that conduct the fastest (35–75 m/s). A-delta (Aδ) fibers are smaller but also myelinated and have slower conduction speeds (>2 m/s). C fibers consist of small, unmyelinated fibers with the slowest conduction speeds (<2 m/s). Aβ fibers typically respond to light touch, pressure, or vibration. Aδ and C-fibers respond to the sensations of pain or temperature, and those fibers that respond specifically to pain are termed nociceptive nerve fibers. Most nociceptive nerve fibers are polymodal, meaning that they respond to all types of stimuli, including mechanical, thermal, and chemical. Nociceptive Aδ and C fibers appear to work in concert by, respectively, providing the early (sharp, intense) and late (burning, throbbing) sensations of acute pain (Meyer and Campbell 1981 #784) (see also Al-Chaer 2011).
When a nociceptive receptor is exposed to a noxious stimulus, its ion channel opens partially depolarizing the nerve ending, a process termed transduction. When a sufficient number of ion channels are opened, an action potential is generated that transmits an afferent signal to the dorsal horn of the spinal cord. All primary afferents use glutamate as their principle neurotransmitter and, therefore, induce excitatory postsynaptic potentials (EPSP). Many activated nociceptors also play an efferent role in pain signal transduction by peripherally releasing neuropeptides like substance P and calcitonin gene-related peptide (CGRP). These neuropeptides induce an inflammatory reaction (vasodilation, plasma extravasation, attraction of macrophages, mast cell degranulation, etc.) termed neurogenic inflammation (Schaible and Richter 2004).
The amount of external stimulation (e.g., heat, pressure, or chemical) that is required to generate an action potential is termed the activation threshold. The role of nociceptive neurons is to identify actual or potential injury, and so their activation thresholds are usually high. However, as will be discussed, the activation threshold of a neuron is not static. In fact, the ability of the nervous system to adjust its neuronal responses to external stimuli is a central concept of neuronal plasticity.
All peripheral afferent fibers direct their action potentials to the dorsal horn, which is the main hub connecting the peripheral nervous system (PNS) and the central nervous system (CNS). At the level of the dorsal horn, a confluence of ascending and descending spinal tracts interact with the central terminals of peripheral afferents. Such bidirectional signaling allows for the nervous system to both process and adjust to somatosensory input almost instantaneously.
The dorsal horn is anatomically divided into six layers or laminae based on the size and packing density of its neurons as identified by Rexed (1952 #782). Peripheral afferent fibers terminate within the laminae of the dorsal horn in an organized fashion according to diameter and type. Most Aδ and C-fiber central terminals are found in the superficial laminae I and II (respectively, called the marginal layer and the substantia gelatinosa) while the larger Aβ-fibers tend to arborize in the deeper laminae III–V. Ascending projection neurons relay sensory input from the dorsal horn to various supraspinal targets where processing of information continues. Descending neurons travel in the opposite direction and allow for various supraspinal regions to influence how easily sensory input flows through the dorsal horn (discussed further below). Interneurons are also found within the dorsal horn. These neurons do not travel outside of the spinal cord and tend to arborize locally. While incompletely understood, at least part of their known role is to modulate sensory throughput at the dorsal horn in response to signaling received from both peripheral afferent fibers and central descending axons.
Ascending projection neurons can be classified as either nociceptor specific (NS) or wide dynamic range (WDR) based on the type of sensory input they receive. NS neurons are predominantly found in lamina I and the outer part of lamina II of the dorsal horn where they receive input from Aδ and C-fiber nociceptive afferents. Most often NS neurons interact with their afferent inputs indirectly via interneurons; however, some direct synapses between Aδ fibers and NS projection neurons in lamina I have been identified (Grudt and Perl 2002 #785). WDR neurons are located deeper in the dorsal horn in lamina III–V. These neurons directly synapse with both low-threshold Aβ and nociceptive Aδ and C-fibers.
WDR neurons have graded responses to the sensory input that they receive so that non-noxious stimuli induce lower impulse firing rates than noxious stimuli. In contrast, NS neurons do not respond at all to non-noxious stimuli, but respond fully to noxious stimuli. While both WDR and NS neurons have spontaneous discharge rates, WDR neurons have faster basal rates (11 Hz) than NS neurons (3–5 Hz).
Although WDR neurons are located primarily in the deeper laminae, they have been found to send dendritic connections to the more superficial laminae of the dorsal horn (Naim et al. 1997 #786). Such neuronal organization supports the gate control theory of pain originally proposed by Melzack and Wall (1965 #787). This theory posits that intrinsic Aβ activity inhibits nociceptive signaling by Aδ and C-fibers. In fact, the ability of such low-threshold non-noxious receptor activity to partially inhibit the conduction of nociceptive signaling at the level of the dorsal horn has been demonstrated (Bini et al. 1984). Furthermore, an established connection between non-noxious and noxious signaling pathways provides a mechanism for how certain chronic pain conditions such as allodynia (pain experienced from non-noxious stimuli) may exist.
Numerous ascending projection tracts extend from the dorsal horn to various targets in the brainstem and higher cortical regions. The destinations of these tracts provide clues into what regions of the brain are involved in pain processing and how other psychological phenomena, such as emotion or attention, may influence pain perception.
The largest and most studied of the ascending projection systems is the spinothalamic tract (Fig. 19.1). Axons of the spinothalamic tract are concentrated in two locations of the spinal cord. The lateral spinothalamic tract travels in the middle of the lateral funiculus, and the medial spinothalamic tract travels in the middle of the anterior funiculus. The lateral spinothalamic tract projects to the somatosensory region of the brain and the medial spinothalamic tract projects to prefrontal targets including the anterior cingulate. Based on these respective destinations, historically these tracts have been associated with the sensory-discriminative and the cognitive-emotional components of pain sensation. However, emerging research continues to blur these lines, especially in the situation of chronic pain (Apkarian et al. 2009).
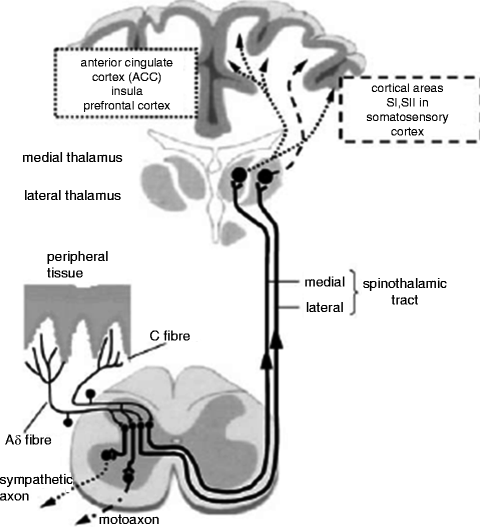
Fig. 19.1
Scheme of the nociceptive system with nociceptive. Free nerve endings in the peripheral tissue, afferent nerve fibers, and their synapses in the dorsal horn of the spinal cord. From the dorsal horn, the ascending tracts including the medial and lateral spinothalamic tracts project to the thalamus. Relay nuclei in the thalamus then project rostrally to the anterior cingulate cortex, insula, prefrontal cortex, and somatosensory cortex. Interneurons also exist in the dorsal horn that serve to modulate secondary synaptic propagation thereby attenuating or facilitating nociceptive signaling. Interneurons have also been found to interact with sympathetic and motor nerves as well (Schaible and Richter 2004)
Other ascending tracts also exist that appear to be as vital, if not even more important, than the spinothalamic tracts regarding the development of chronic pain. Projections to the catecholamine cell groups (A1–7) are thought to be involved with the autonomic responses (Ex. tachycardia, vasoconstriction) associated with pain. The parabrachial nucleus also receives sensory input from the spinal cord that may allow it to play a role in integrating visceral and peripheral nociceptive sensation. The fact that the parabrachial nucleus projects to both the hypothalamus and reticular formation suggests that it may be involved with the neuroendocrine and arousal responses to pain. The parabrachial nucleus’ interactions with the amygdala provide a potential pathway connecting pain and emotion (Price et al. 2006).
As will be discussed in greater detail (see also, Naylor et al. 2011) a variety of cortical regions appear to be involved in the higher processing of pain. For acute pain, the major contributing brain regions are the primary and secondary somatosensory, insular, anterior cingulate, and prefrontal cortices. Many of these structures have been found to receive projections both directly from the spinal cord and indirectly via the spinothalamic pathways (Price et al. 2006 #801). There is also growing recognition that in chronic pain states the relative contributions of each of these regions and the timing of their interactions with each other changes. Furthermore, patients with chronic pain engage regions of the brain that usually remain silent in healthy patients experiencing acute pain. Many of these newly engaged regions are heavily involved in cognitive and emotional functioning, providing a mechanism for the strong affective component of chronic pain that is so often seen in clinical pain population (Apkarian et al. 2005 #798, 2009 #794).
Electrophysiological Concepts of Neuroplasticity
Exposure to noxious stimuli promotes neuroplastic changes that clinically result in hyperesthesia. Hyperesthesia is an increased sensitivity to stimulation, both painful and nonpainful. Hyperesthesia is a broad term that includes several different types of painful sensations (Merskey and Bogduk 1994):
Hyperalgesia is an increased response to a stimulus that is normally painful.
Allodynia is a painful response to a stimulus that is normally not painful.
Dysesthesia is an unpleasant abnormal sensation that may be either spontaneous or evoked.
While pain is a subjective phenomenon, the process of nociception (the encoding and processing of noxious stimuli) is an objective phenomenon that can be measured via electrophysiological testing. Electrophysiologically, hyperesthesia is observed as increased nerve fiber activity following a noxious stimulus and is termed sensitization. Sensitization occurs when receptors display a decreased activation threshold for stimulation, an increased response to suprathreshold stimuli, and spontaneous electrophysiological activity (Meyer et al. 2006). The relationship between the electrophysiological findings observed with sensitization and patients’ subjective experiences with hyperesthesia is detailed in Table 19.2.
Table 19.2
Comparison of characteristics of hyperesthesia and sensitization
Hyperesthesia (subjective response) | Sensitization (electrophysiologically recorded nerve fiber response) |
|---|---|
Increased pain to suprathreshold stimuli (hyperalgesia) | Increased electrical response to suprathreshold stimuli |
Decreased pain threshold (allodynia) | Decreased activation threshold for electrical response |
Spontaneous pain (dysesthesia) | Spontaneous electrical activity |
An individual nociceptor innervates a specific area on the skin that can be measured objectively. This area of nociceptive coverage is termed the receptor field. When a nociceptor is activated by a noxious stimulus, the receptor field of that nociceptor becomes hyperalgesic, thus forming the zone of primary hyperalgesia. Neuroplastic changes primarily occurring in the PNS, or peripheral sensitization, form the zone of primary hyperalgesia. However, after a noxious stimulus, the area outside of the original receptor field can also become hyperalgesic. This surrounding area is termed the zone of secondary hyperalgesia, and it develops as a result of neuroplastic changes occurring in the CNS, or central sensitization (Fig. 19.2) (Joshi and Ogunnaike 2005).
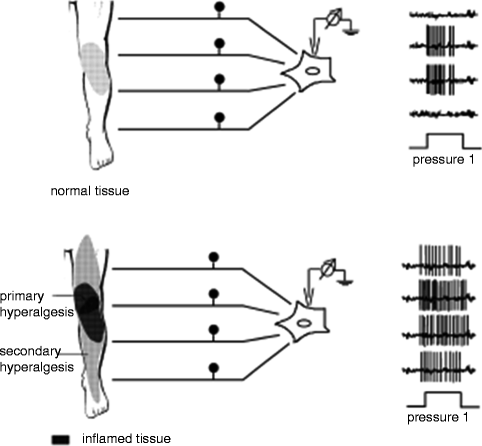
Fig. 19.2
When a noxious stimulus is applied to tissue, peripheral sensitization results in the zone of primary hyperalgesia. As result of central sensitization, the expanded zone of secondary hyperalgesia is formed (Schaible and Richter 2004)
That different mechanisms are involved in the development of the primary and secondary zones of hyperalgesia were elegantly demonstrated in an experiment by LaMotte et al. (1991). Capsaicin (a substance found in hot peppers that directly activates nociceptors producing both zones of primary and secondary hyperalgesia) was injected subcutaneously into an upper extremity that had already received a proximal nerve block, sparing the CNS of nociceptive input. By creating this interruption between the peripheral and CNS, formation of the zone of secondary hyperalgesia was entirely inhibited supporting the CNS’s integral involvement in this process (LaMotte et al. 1991).
However, during surgery, nerves not only relay nociceptive input resulting from surrounding tissue injury but also suffer injury themselves. Such injury to nervous tissue results in a specific type of pain called neuropathic pain. Characteristics of neuropathic pain can be divided into negative and positive symptoms. Negative symptoms include the sensory and motor deficits that result from the injury. Positive symptoms include any allodynia, hyperalgesia, dysesthesia, or hyperpathia at the site of the nerve injury. Hyperpathia refers to a combination of dulled sensation with increased stimulus threshold and poor localization; extreme pain with repeated stimulation that persists after the stimulation has ended; and spread of the pain from the site of stimulation. Hyperpathia appears to be unique to neuropathic pain (Devor 2006).
Peripheral sensitization and the neuropathic pain resulting from nerve injury work in conjunction to send a barrage of nociceptive signaling to the dorsal horn. This massive influx of nociceptive input, in turn, causes central sensitization to occur which further amplifies what is relayed to the brain. Higher processing then incorporates behavioral and historical components such as emotion, past memories, and psychopathological conditions into received sensory information allowing these elements to also modify how pain is perceived. Furthermore, the brain utilizes the descending tracts to exert its influence over pain processing at the level of the dorsal horn as well. Some of the mechanisms thought to be responsible for the above processes will be discussed below.
Peripheral Sensitization
The sensitivity of the peripheral nociceptors increases with repeated stimulation. Heat sensitivity and thermal hyperalgesia are at least partly mediated by the capsaicin-sensitive vanilloid 1 receptor/ion channel complex (TRPV1). Increased intracellular calcium levels that occur as a result of repeated activation are believed to cause conformational changes increasing sensitivity of the receptor (Guenther et al. 1999). Mechanoreceptors are thought to open their associated ion channels in response to external pressure allowing cations to depolarize the nerve terminal, achieve activation threshold, and initiate an action potential. Swelling secondary to inflammation may allow these channels to more effectively open, thereby, increasing their sensitivity (Belmonte and Cervero 1996). Furthermore, some nociceptive fibers increase their receptive fields after activation, resulting in a greater response to stimulus ratio, which can produce more pain (Reeh et al. 1987; Thalhammer and LaMotte 1982).
Numerous inflammatory mediators are released upon exposure to a noxious stimulus or tissue injury including bradykinin, histamine, serotonin, adenosine derivatives, arachidonic acid metabolites, and various cytokines (Fig. 19.3). Some of these substances directly activate nociceptors further increasing their sensitivity by means of repeated stimulation. However, other mediators initiate modulatory changes to the PNS by initiating intracellular cascades leading to increased nociceptor sensitivity (Bhave and Gereau 2004; McMahon et al. 2006). Bradykinin both directly activates nociceptors as well as sensitizes the capsaicin receptor (TRPV1) via a protein kinase C pathway (Okuse 2007). Adenosine triphosphate (ATP) released from injured tissue appears to bind to its associated ion channel, P2X3, increasing calcium influx into nociceptive nerve fibers. Pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), IL-6, and the chemokine IL-8 have been shown to produce mechanical and thermal hyperalgesia following injection. However, the cytokines also appear to induce peripheral sensitization via phosphorylation of nociceptive receptors by means of various intracellular kinase pathways. The prostaglandins phosphorylate the tetrodotoxin-resistant sensory neuron specific (TTX-SNS) sodium ion channel, an ion channel specific to Aδ and C-fiber nerves, via a protein kinase A (PKA) or C pathway reducing its activation threshold (Cesare et al. 1999; Khasar et al. 1999).
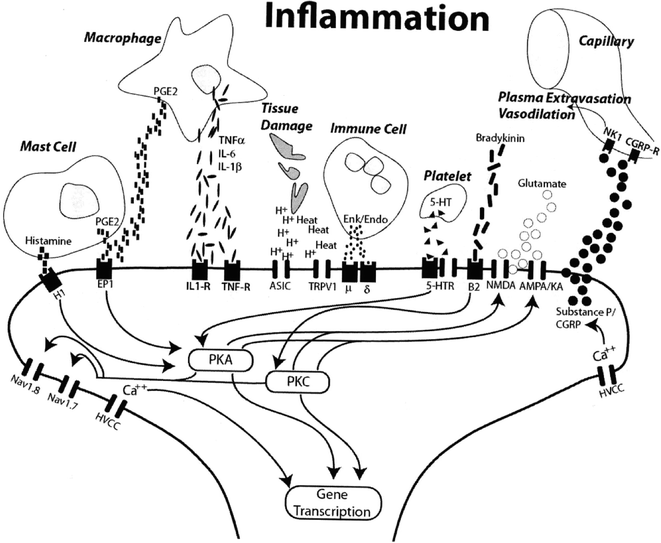
Fig. 19.3
Inflammatory mediators released in response to a noxious stimulus both directly activate some receptors as well as initiate a series of intracellular cascades that result in both post-translational receptor modulation and gene transcription modification, all of which result in peripheral sensitization (Sluka 2009)
Additionally, several pro-inflammatory mediators have been shown to recruit a special group of nociceptive nerve fibers, termed mechanically insensitive afferents (MIAs). These fibers, as their name suggests, are normally unresponsive to mechanical stimuli and not involved in nociceptive transmission. However, upon activation, they become profoundly sensitive even to subthreshold stimuli that would normally not be noxious. This finding correlates with the hypersensitivity noted immediately following an injury (Davis et al. 1993). A cyclic AMP/PKA second messenger cascade has been implicated as the mechanism by which recruitment of MIA receptors occurs (Levy and Strassman 2002).
Inflammation also releases neurotrophins such as nerve growth factor (NGF) into the involved tissue. Neurotrophins are a family of proteins that normally act to induce survival, development, and function of neurons; however, when their concentration levels are increased they function to produce a hyperalgesic response. NGF directly causes degranulation of mast cells, and as a result provides a positive feedback loop further increasing the amount of inflammatory mediators available to induce sensitization of the peripheral nociceptors. NGF also modifies gene expression by binding to receptor tyrosine kinase A (trkA). This interaction upregulates synthesis of SNS, TRPV1 channels, substance P, calcitonin-gene-related peptide (CGRP), and bradykinin receptors further contributing to peripheral sensitization (Michael and Priestley 1999; Okuse 2007; Schaible and Richter 2004; Tate et al. 1998).
Peripheral Nerve Injury
Injury to a peripheral nerve results in neuropathic pain. While healthy nerves tend to only fire after being stimulated, injured nerves often exhibit abnormal, spontaneous electrical activity termed ectopy (Fukuoka et al. 2000; Wu et al. 2001). These ectopic discharges involve not only the smaller Aδ and C-fiber nerves, but also the larger myelinated Aβ-fibers that typically encode innocuous mechanosensory information.
Ectopy develops because nerve injury induces internal changes that lead to a hyperexcitable state. As injured nerves undergo Wallerian degeneration, an inflammatory milieu develops causing increased levels of NGF to be expressed. NGF, in turn, induces changes in membrane ion channel expression resulting in both increased channel density as well as altered channel kinetics. A higher density of ion channels promotes ectopy by increasing the likelihood that depolarization occurs (Aurilio et al. 2008).
The specific role of ion channels in the development of ectopy is worth addressing. Nine subtypes of sodium ion channels have been identified and classified by their respective sensitivity or resistance to the neurotoxin, tetrodotoxin (TTX). The TTX-sensitive sodium channels become inactive quickly, resulting in burst-like signal transmissions that are most consistent with the electrical activity of ectopy. The TTX-resistant sodium channels which have slower inactivation and activation kinetics are most concentrated on the smaller Aδ and C-fiber neurons. Following a nerve injury, both TTX-sensitive and resistant channels are upregulated at the damaged site as well as along the neuron. These changes are thought to alter the membrane properties of the neuron, so that rapid firing rates (bursting ectopic discharges) are produced (Woolf and Mannion 1999). The ability of the intrinsically slower TTX-resistant channels to produce ectopic activity is believed to be a result of cascade mediated conformational changes that increase channel conductivity (Aurilio et al. 2008). In addition to the abnormal accumulation of sodium channels, there is evidence that nerve injury also promotes an increased number of calcium channels to be expressed that may contribute to ectopic activity (Xie et al. 1993). These specific phenotypic changes are important to understand as they may offer routes for targeted pharmocotherapy in the future. As will be discussed later, many of the medications used to treat neuropathic pain are nonspecific and, therefore, have many side effects. Developing agents with high affinities specific to these ion channels is a high priority in the pharmaceutical industry.
Normally, the sympathetic nervous system is not involved in nociception. However, some axons begin to express α-adrenoceptors following nerve injury, causing them to become sensitive to circulating catecholamines and norepinephrine released by postganglionic sympathetic terminals. In such a manner, the sympathetic nervous system may also become a source of nociceptive transduction, termed sympathetically maintained pain (Woolf and Mannion 1999).
Central Sensitization
Central sensitization can be viewed as having two major components. First, it serves to amplify any sensory input received at the dorsal horn. Second, it converts innocuous sensory input from non-nociceptive afferents into nociceptive transmission. In addition to CPSP, central sensitization is also involved in the development of neuropathic pain (diabetic neuropathy, postherpetic neuralgia), inflammatory pain (rheumatoid arthritis, lupus), migraine, irritable bowel syndrome (IBS), and fibromyalgia. Neurons in the dorsal horn that have been affected by central sensitization exhibit some or all of the following (Latremoliere and Woolf 2009):
Development of or increases in spontaneous activity
A reduction in the threshold for activation by peripheral stimuli
Increase in the responsiveness to any suprathreshold input
Expansion of the extent of the receptive fields of dorsal horn neurons
When inflammatory and neuropathic pain in the periphery conduct a barrage of nociceptive signaling to the spinal cord, neuroplastic changes resulting in central sensitization begin to occur. Normal nociceptive signal conduction involves the primary afferent neuron releasing the excitatory neurotransmitters, glutamate, and aspartate into the synaptic cleft. These neurotransmitters then activate the alpha-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid (AMPA) receptors on the postsynaptic membrane allowing for signal propagation to continue rostrally. However, when AMPA receptors are continuously activated, prolonged depolarization of the postsynaptic membrane occurs allowing for a normally quiescent N-methyl-d-aspartate (NMDA) channel to become activated through a loss of static inhibition by magnesium ions (Mayer et al. 1984). Activation of the NMDA receptor creates a surge in intracellular calcium which, in turn, activates numerous intracellular pathways that contribute to the maintenance of central sensitization. Together, the AMPA and NMDA receptors amplify the incoming nociceptive signal throughput in a process termed windup.
Additionally, depolarization triggers the release of neuropeptides, including substance P (SP) and CGRP, into the dorsal horn synapse. These neuropeptides activate G-protein coupled receptors, which slow postsynaptic depolarization, further allowing temporal summation and amplification of the nociceptive signal to occur. SP binds to its neurokinin 1 (NK-1) receptor activating a PKC pathway that phosphorylates the NMDA receptor. Phosphorylation of the NMDA receptor also stabilizes the receptor in its active state (Woolf and Salter 2006). These neuropeptides reside in vesicles located in the presynaptic terminal. When depolarization occurs, voltage sensitive calcium channels (VSCC) open increasing intracellular calcium concentrations, thereby allowing the vesicles to fuse with the presynaptic membrane and release their contents. Of the various types of calcium channels expressed in the nervous system, evidence suggests that the high-threshold N-type channels located in the superficial Rexed laminae (discussed further below) are most important to the development of central sensitization (Fig. 19.4) (Aurilio et al. 2008).
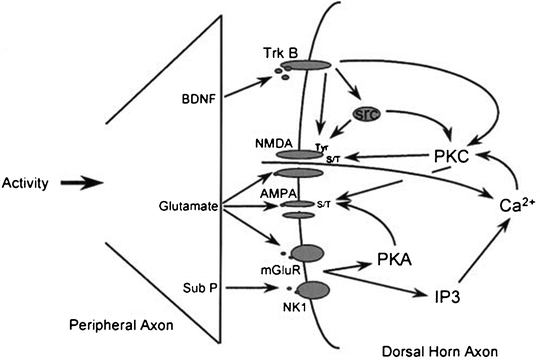
Fig. 19.4
Repeated barrage of nociceptive signaling at the dorsal horn results in activation of the NMDA receptor as well as initiation of multiple intracellular cascade pathways leading to post-translational and transcriptional changes (Woolf and Costigan 1999)
Synaptic connections at the dorsal horn are not permanent, and their rearrangement may explain how normally innocuous input from mechanosensory Aβ-fibers is perceived as painful after central sensitization has occurred. Typically, nociceptive fibers synapse superficially in the dorsal horn at Rexed laminae I and II while mechanosensitive fibers synapse at the deeper Rexed laminae. Aβ-fibers do have dendritic extensions to nociceptors in the more superficial laminae. Normally signals via these extensions are thought to be inhibited by interneurons. One theory of allodynia is that a loss of this inhibition occurs allowing for non-noxious stimuli to be miscoded as a nociceptive signal (Todd 2010 #817). Furthermore, following central sensitization the synaptic locations of the Aβ-fibers migrate superficially so that they activate secondary neurons of the nociceptive pathway (Schaible and Richter 2004). Such rearrangement of synaptic connections at the dorsal horn may extend rostrally or caudally one or more levels contiguously.
Other factors may also have roles in the development of secondary hyperalgesia. For example, tissue damage itself, particularly to low-threshold non-noxious peripheral receptors, may also facilitate this process. The normal activity of these innocuous receptors partially inhibits the conduction of pain signals upon activation of nociceptors (Bini et al. 1984). Therefore, loss of this innocuous transmission may result in a central disinhibition of nociceptor input (Meyer et al. 2006). As will be discussed in the next section, supraspinal influence at the level of the dorsal horn is also very much involved in the development of central sensitization.
Descending Tracts
The ability of the brain to modulate nociceptive signaling at the level of the dorsal horn is dependent on two structures: the PAG and the rostral ventromedial medulla (RVM). The PAG is located in the midbrain and receives direct inputs from numerous higher structures including the anterior cingulate, insular cortex, and amygdala. Indirectly, structures such as the nucleus accumbens and lateral hypothalamus project to the PAG. The PAG also interacts with its neighboring regions, the nucleus cuneiformis, the reticular formation, the locus coeruleus, and other brainstem catecholaminergic nuclei. Finally, the PAG receives direct projections from lamina I nociceptive neurons.
The PAG exerts its influence over nociceptive processing at the dorsal horn via its interaction with the RVM. The RVM is located in the medulla and includes the midline nucleus raphe magnus and the adjacent reticular formation that lies ventral to the nucleus reticularis gigantocellularis. The major descending tracts to the dorsal horn of the spinal cord initiate from the RVM. Three classes of neurons that travel from the RVM to the dorsal horn have been identified: on-cells, off-cells, and neutral cells. These neurons are found throughout the RVM and are not anatomically separable; they have instead been classified based on their activity responses to a noxious stimulus. Off-cells display an abrupt pause in ongoing activity immediately following a noxious stimulus and are believed to contribute to the inhibitory influences of the RVM on the dorsal horn. On-cells display a burst of activity immediately following a noxious stimulus and are believed to contribute to the facilitatory influences of the RVM on the dorsal horn. Neutral cells show no change in activity related to noxious stimuli (Fields et al. 2006 #810).
While the PAG-RVM descending modulatory system is most known for the attenuating effects it has on pain, more recently there has been growing evidence that it may have significant roles in facilitating chronic pain states as well. Low-intensity electrical stimulation of the RVM or direct injection of the neurotransmitter glutamate has been shown to increase spinal nociception. These facilitatory influences appear to involve an anatomically separate descending projection system in the ventrolateral funiculi and to be mediated by spinal serotonin and cholecystokinin receptors. Furthermore, secondary hyperalgesia following certain pain states such as mustard oil-induced sensitization and neuropathic pain after spinal nerve ligation can be attenuated by inactivation of the RVM. NMDA, neurontensin (NT), and nitric oxide (NO−) receptors in the RVM appear to be involved in these processes. An illustration of this proposed spinobulbospinal pathway and its role in secondary hyperalgesia is presented in Fig. 19.5 (Urban 1999 #814).
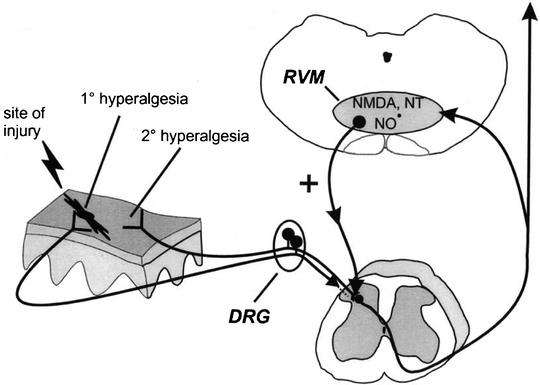
Fig. 19.5
Summary diagram illustrating a significant supraspinal contribution to secondary, but not primary, hyperalgesia. As will be discussed further, peripheral injury results in activation and sensitization of peripheral nociceptors and subsequent enhanced excitability of dorsal horn neurons (central sensitization) that contributes to primary hyperalgesia (at the site of injury) and secondary hyperalgesia (adjacent/distant from the site of injury). Additionally, it has been proposed that stimulation of nociceptors activates a spinobulbospinal loop, engaging centrifugal descending nociceptive facilitatory influence from the RVM. Facilitatory influences are activated by NMDA, NO−, and NT receptors in the RVM and descend to multiple spinal segments to contribute significantly to secondary hyperalgesia. In contrast, primary hyperalgesia does not involve descending facilitatory influences from supraspinal sites and is likely the direct result of peripheral nociceptor sensitization and neuroplasticity intrinsic to the spinal cord. For clarity, the afferent input to the spinal dorsal horn from the site of injury is illustrated as not entering the spinal cord (which it certainly does) (Urban 1999 #814)
In its healthy state, the basal tone of this processing appears to be mostly inhibitory to prevent spontaneous nociceptive signals from occurring in the absence of actual noxious stimuli or tissue injury. This inhibitory state does not appear to opioid driven given the fact that intrathecal injection of the opioid inhibitor naloxone does not cause hyperalgesia. Serotonin has been proposed as partially responsible for the tonic inhibitory state. Supporting this hypothesis are the facts that the RVM is the exclusive source of serotonin in the dorsal horn, serotonergic RVM neurons continuously slowly release serotonin into the dorsal horn, and intrathecal administration of serotonin antagonists has been found to reduce the effectiveness of the RVM to produce analgesia. However, as mentioned above at least one serotonin receptor (5-HT3) appears to also be involved in facilitating certain pain states, suggesting that serotonin probably plays a more complex role in modulating pain (Fields et al. 2006 #810).
Behavioral Factors in Neuroplasticity
Certain behavioral states have consistently been shown to alter pain perception. Studies evaluating the relationship of attention and pain have reliably demonstrated that when subjects are distracted they report less pain (Levine et al. 1982 #824; Miron et al. 1989 #826). This is obviously a difficult association to unravel because pain itself is an attention-demanding phenomenon. In fact, a decreased ability to distract attention away from pain may be a feature common to chronic pain patients (Grisart and Plaghki 1999 #827).
The pathways and mechanisms behind how attention modifies pain perception are still incompletely understood. Neurophysiological studies have shown that modulation from attentional changes occurs even at the level of the dorsal horn (Villemure and Bushnell 2002 #822). The aid of functional MRI (fMRI) has also demonstrated that attention influences many of the same brain regions involved with pain processing. Tracey et al. observed that PAG activity was significantly increased when subjects were distracted from pain (Tracey et al. 2002 #829). Other areas of the brain involved in attention and pain appear to be the secondary somatosensory cortex (S2), anterior cingulate cortex, and insula (Sawamoto et al. 2000 #836).
The inability of chronic pain patients to distract themselves from the experience of pain has been proposed as one driving maladaptive behavior. Normally, the pain associated with repeated exposure to a noxious stimulus diminishes over time in a process termed habituation or a time-dependent decline of vigilance. The same areas of the brain thought to be crucial for maintaining vigilance are also involved in focusing attention to pain. Therefore, patients with chronic pain may exhibit a “hypervigilant” state in which they fail to return to their basal sensory states and disengage pain (Lorenz and Tracey 2009 #838).
Another central concept to understanding the relationship between behavior and pain is that of placebo and nocebo behavior (see also Pollo and Benedetti 2011). A placebo effect is the concept that the expectation of pain relief is able to independently provide analgesia. Mechanistically, this process is thought to be driven by the prefrontal cortex via the PAG-RVM descending modulatory system. In contrast to a placebo effect, a nocebo effect is that of increased pain secondary to a negative expectation of pain. fMRI has associated the S2 and posterior insula with the nocebo effect, and there is evidence that the hyperalgesia is mediated by spinal cholecystokinin. Such maladaptive behaviors such as anxiety, catastrophizing, and excessive fear of injury are believed to evoke nocebo phenomenon. Key brain regions and how they are thought to relate to behavior and pain can be seen in Fig. 19.6 (Lorenz and Tracey 2009 #838).
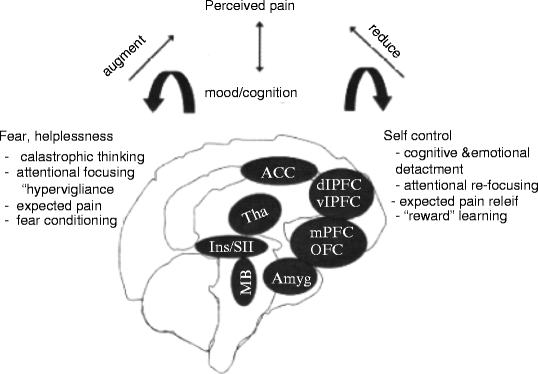
Fig. 19.6
Summary figure of main brain regions determined to be relevant in the psychological amplification and reduction of the pain experience based on bodily or psychological context. ACC anterior cingulate cortex; Amyg amygdala; dlPFC dorsolateral prefrontal cortex; Ins insular cortex; MB midbrain; mPFC medial prefrontal cortex; OFC orbitofrontal cortex; SII secondary somatosensory cortex; Tha thalamus; vlPFC ventrolateral prefrontal cortex (Lorenz and Tracey 2009 #838)
Risk Factors and Preventative Therapies
The pathophysiologic changes underlying CPSP are complex, and treatment after it has developed is often underwhelming. Hence, there has been a greater focus on preventative therapy. The most obvious way to prevent the development of CPSP is to not undergo the surgery. Yet, a reduction in the number of surgeries would still only partially solve the problem. No matter how selective one is, many patients ultimately require surgery, especially in the field of palliative care. Therefore, a better understanding of the predisposing risk factors, both procedurally as well as biopsychosocially, is required in order to help design a successful preventative therapeutic strategy.
Procedural
Ample evidence exists demonstrating that a given surgical approach can influence the development of CPSP. The major cause of CPSP is thought to be nerve injury, either directly from the surgical insult or from other consequences such as inflammation or scar tissue. Thus, new surgical techniques have been designed to reduce the overall amount of tissue and nerve injury, and in doing so, to help prevent CPSP. Laparoscopically assisted surgery has been shown to result in decreased long-term post-surgical pain for both hernia repair and cholecystectomy (Grant et al. 2004; Stiff et al. 1994). Nerve sparing approaches for both mastectomy and thoracotomy have also reduced post-surgical morbidity (Cerfolio et al. 2003; Jung et al. 2003). Surgical techniques such as sentinel lymph node biopsy, which were developed to reduce the number of axillary radical dissections performed, have also decreased the incidence of chronic postmastectomy pain (Sclafani and Baron 2008).
An increased risk of CPSP has been associated with a surgical duration of greater than 3 h (Peters et al. 2007). This is probably because a longer, more complicated procedure is likely to involve greater amounts of tissue injury and nerve damage. Furthermore, the incidence of CPSP has been observed to be decreased when the surgery is performed at a high vs. low volume surgical unit (Tasmuth et al. 1999). This finding is commensurate with several studies reporting a favorable association between high procedural volume centers and reduced surgical morbidity (Battaglia et al. 2006; Rectenwald and Upchurch 2007; Wilt et al. 2008).
Concomitant treatments may also increase the incidence of CPSP. Chronic pain has been reported to be increased following amputation and mastectomy when performed in close conjunction with radiation or chemotherapy (Gulluoglu et al. 2006; Smith and Thompson 1995). Whether such therapeutic regimens contribute to pain following other types of surgery is still uncertain; however, the potential for radiation or chemotherapy alone to cause neuropathic pain is also well documented (Argyriou et al. 2008; Forman 1990).
Biopsychosocial
Acute pain appears to correlate with the development of chronic pain. This relationship exists for acute pain in both the preoperative and postoperative settings. Amputees with severe preoperative pain are more likely to develop intense phantom limb pain than those who report no or only mild preoperative pain (Jensen et al. 1985; Nikolajsen et al. 1997). Similarly, severe postoperative pain has been shown to be an independent risk factor for the development of CPSP after thoracotomy (Katz et al. 1996), mastectomy (Poleshuck et al. 2006), herniorrhaphy (Aasvang and Kehlet 2005), and Cesarean section (Nikolajsen et al. 2004). These findings are consistent with the idea that neuroplastic changes begin in the acute setting.
While the true gender prevalence of chronic pain remains debated, women report more severe pain, more frequent bouts of pain, and more anatomically diffuse pain compared to men (Hurley and Adams 2008, see also Keogh 2011). This tendency for the female population to report greater pain has been observed with postthoracotomy pain (Gotoda et al. 2001). Younger age may also predispose the development of CPSP. Poleshuck et al. reported that the probability of developing chronic pain after breast surgery decreased 5% for each additional year of age (Poleshuck et al. 2006). This relationship may be partly explained by younger patients tending to have more aggressive and advanced disease, therefore, requiring more extensive surgery (Kroman et al. 2000).
Conflicting evidence exists regarding psychological predictors for CPSP. Harden et al. found that preoperative anxiety was related to the development of complex regional pain syndrome (CRPS) following total knee arthroplasty (Harden et al. 2003). However, in another study by Katz et al., no relationship was found between depression and anxiety and postthoracotomy pain (Katz et al. 1996). It should be noted, however, that Katz’s study questioned patients 1 day prior to surgery, and this timing may have falsely elevated the number of patients expressing symptoms of both depression and anxiety. Traits of neuroticism and an introverted personality have been found to be related to postcholecystectomy pain (Borly et al. 1999). A recent review of the literature by Hinrichs-Rocker et al. (2009) observed that depression, psychological vulnerability, and stress to be the most reliable psychological predictors of CPSP currently known.
Pain catastrophizing may also be related to the development of CPSP. Pain catastrophizing is defined as an exaggerated negative orientation to aversive stimuli that involves rumination about painful sensations, magnification of the threat value of the painful stimulus, and perceived inability to control pain (Granot and Ferber 2005). These inappropriate pain behaviors are generally considered maladaptive coping strategies, and as such, often result in increased anxiety levels. Pain catastrophizing has been demonstrated to be a risk factor for increased chronic pain in rheumatoid arthritis patients (Keefe et al. 1989). Currently available studies evaluating the role of catastrophizing in CPSP, however, are conflicting. While catastrophizing has been shown to predict acute post-surgical pain intensity (Pavlin et al. 2005), other studies have been unable to establish such a correlation with CPSP, despite the fact that severe acute post-surgical pain is itself a risk factor for CPSP (Hanley et al. 2004; Peters et al. 2007). In fact, more frequent catastrophizing 1 month postoperatively was found to be associated with greater improvement in the pain score at 2 years postoperatively. This finding may be due to this study having only evaluated the absolute change in pain scores as opposed to actual pain scores. Therefore, a patient with frequent catastrophizing at 1 month may have a higher overall pain score, thereby giving that patient more room to improve. However, one alternative explanation provided by the authors was that early catastrophizing has a paradoxically beneficial role by prompting earlier recognition and treatment.
A patient’s social environment as a predictor of CPSP has also been assessed. One study investigating the risk factors for phantom limb pain found lack of social support to be a major predictor (Jensen et al. 2002). This finding was supported in a subsequent study evaluating the incidence of postamputation pain (Hanley et al. 2004). Solicitous behavior, in which family members (often unknowingly) reinforce a patient’s pain behaviors, may also be a risk factor for developing CPSP (Katz and Seltzer 2009).
Finally, there may be genetic predisposition to the development of chronic neuropathic pain that increases the likelihood of CPSP. The link has been demonstrated in animal studies, in which specific strains of mice are more likely to develop neuropathic pain after nerve injury (Seltzer et al. 2001). There is also evidence that patients with CPSP are more likely to have other functional pain syndromes such as IBS, temporomandibular joint disorder (TMJD), migraine, and fibromyalgia (Macrae 2008). Yarnitsky et al. tested patients’ diffuse noxious inhibitor control (DNIC), a marker of one’s overall endogenous analgesia system, prior to undergoing thoracotomy. Although the level of DNIC did not correlate with acute post-surgical pain, it was directly related to the development of chronic postthoracotomy pain. This finding suggests that not only is there likely a genetic predisposition to developing CPSP, but also that the mechanisms of acute and CPSP are most likely distinct (Yarnitsky et al. 2008).
Preventative Therapies
The nociceptive barrage that is thought to trigger the peripheral and central sensitization that ultimately causes CPSP may occur at any time point in the perioperative period – preoperative, intraoperative, and postoperative. However, CPSP is different than other forms of chronic pain in that the timing of the noxious stimulus (the surgery insult) is usually known prior to its occurrence; therefore, an opportunity may exist to proactively disrupt the chain of events that leads to CPSP in the face of severe preoperative pain. The concept of preemptive analgesia holds that the application of analgesic techniques prior to the surgical insult will block the central transmission of pain signals, thereby preventing or reducing the neuroplastic changes thought most responsible for the development of CPSP. While several animal studies have supported the role of preemptive analgesia, the ability to reliably reproduce these results in clinical situations has not been achieved (Dougherty et al. 1992; Haley et al. 1990; Yamamoto and Yaksh 1991).
Multiple reasons have been posited to explain this lack of translation from animal to human studies. As mentioned above, the presence of presurgical pain may be sufficient to independently trigger CPSP, despite adequate nociceptive suppression during the intraoperative and postoperative periods. While such preoperative pain is absent in animal studies, it is often present in human clinical settings. Additionally, animal studies typically choose a surgical procedure involving well-defined nociceptive afferents such as the tail or an extremity so that adequate nociceptive suppression can be easily achieved. However, in human studies, the nociceptive inputs are often diffuse, and complete blockade of algesic signaling is much more difficult. Surgeries in animal studies are often shorter than in comparative human study. Such brevity better ensures adequate nociceptive blockade throughout the duration of the procedure. Animal models also lack the psychological factors that have been found to greatly influence the occurrence of CPSP in humans (Aida 2005).
Related posts:
 Educating Patients and Caregivers About Pain Management: What Clinicians Need to Know
Palliative Care Clinical Trials: Generalizability and Applicability in Hospice and Palliative Care Practice
Educating Patients and Caregivers About Pain Management: What Clinicians Need to Know
Palliative Care Clinical Trials: Generalizability and Applicability in Hospice and Palliative Care Practice
 Pain Assessment Tools in Palliative Cancer Care
Pharmacogenetics of Pain: The Future of Personalized Medicine
CAM in Chronic Pain and Palliative Care
Pain Assessment Tools in Palliative Cancer Care
Pharmacogenetics of Pain: The Future of Personalized Medicine
CAM in Chronic Pain and Palliative Care
 Pain Imaging
Pain Imaging

Full access? Get Clinical Tree


