Chapter 26 Acute respiratory failure in chronic obstructive pulmonary disease
The terms ‘chronic obstructive pulmonary or airways disease’ (COPD or COAD) are applied to patients with chronic bronchitis and/or emphysema. COPD affects 5% of the adult population, is the fifth most common cause of death worldwide and is the only major cause of death that is increasing in prevalence.1 Despite this, when an acute deterioration occurs, most precipitating factors are reversible and the outcome is usually good.2 This justifies aggressive management in the majority of patients.
AETIOLOGY
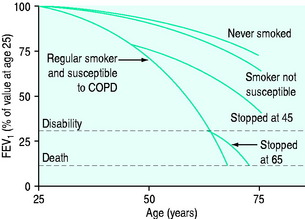
The causes of COPD can be divided into environmental and host factors. Environmental factors include tobacco smoke, air pollution, indoor fumes (e.g. indoor cooking with solid biomass fuel) and poor socioeconomic status. The biggest single factor in over 95% of patients with COPD is tobacco smoking (Figure 26.1). However, only approximately 15% of smokers develop COPD. Marijuana smoking may cause premature and quite advanced bullous emphysema compared with tobacco smokers due to extremely hot and toxic inhaled smoke held at peak inspiration for prolonged periods of time.3 Host factors are the balance between circulating proteases and antiproteases (e.g. alpha-1 antitrypsin deficiency) and the intake of antioxidant vitamins (A, C, E).4
PATHOPHYSIOLOGY
Pulmonary hyperinflation has both static and dynamic components. The static component remains at the end of an expiratory period long enough for all expiratory airflow to cease (30–120 s), enabling the lungs and chest wall to reach their static functional residual capacity (FRC). This component of hyperinflation is due to loss of parenchymal elastic recoil, chest wall adaptation5 and airway closure that occurs throughout expiration. Dynamic pulmonary hyperinflation is the further increase in hyperinflation due to slow expiratory airflow not allowing completion of expiration before the arrival of the next breath. The extent of dynamic hyperinflation depends on the severity of airflow obstruction, the amount inspired (tidal volume) and the expiratory time.6 Thus, the degree of hyperinflation may vary in a patient with changes in minute ventilation due to changes in CO2 production (depending on exercise, diet or the metabolic response to illness) or dead space, as well as with changes in airflow obstruction during an exacerbation.
Central respiratory drive may also be impaired, or poorly responsive to physiological triggers – hypoxaemia or hypercapnia – and lead to chronic hypercapnia. This may occur in the setting of sleep (i.e. obstructive sleep apnoea), obesity or drugs (sedatives, antiepileptics, alcohol).
CHRONIC BRONCHITIS OR EMPHYSEMA?
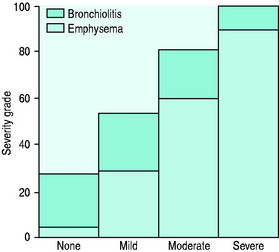
The value of labelling patients as chronic bronchitis or emphysema is uncertain as the two disease processes usually coexist and the principles of management are similar. Five pathophysiologic processes may be present to varying degrees in each patient with COPD: (1) inflammatory airway narrowing (bronchiolitis); (2) loss of connective tissues tethering airways; (3) loss of alveoli and capillaries; (4) hyperinflation; and (5) increased pulmonary vascular resistance. Early/mild COPD tends to be dominated by bronchiolitis with a minimal component of emphysema (Figure 26.2), whereas when COPD becomes severe, the reverse is true. However, recognition that COPD is dominated by one of these patterns is helpful with regard to clinical pattern and prognosis.
CLINICAL FEATURES OF ACUTE RESPIRATORY FAILURE IN COPD
Acute respiratory failure (ARF) in COPD can present with two distinct clinical patterns7 (Table 26.1).
Table 26.1 Clinical differences between normocapnic and hypercapnic chronic obstructive pulmonary disease
| Normocapnic (PaCO2 35–45 mmHg) | Hypercapnic (PaCO2 > 45 mmHg) |
|---|---|
| Emphysema > chronic bronchitis | Chronic bronchitis > emphysema |
| Thin | Obese |
| Pursed-lip breathing | Central nervous system depression: consider the role of oxygen therapy |
| Accessory muscle use | Alcohol, sedatives, analgesics |
| Hyperinflated | Sleep-related hypoventilation |
| Right heart failure late | Right heart failure early |
PRECIPITANTS OF ACUTE RESPIRATORY FAILURE
In approximately 50% of patients, there is an infective cause, in 25% heart failure and in the remaining 25% retained secretions, air pollution, coexistent medical problems (e.g. pulmonary embolus, medication compliance or side-effects) or no cause can be identified8 (Table 26.2).
Table 26.2 Precipitants of acute respiratory failure in chronic obstructive pulmonary disease
| Infective (including aspiration) |
| Left ventricular failure (systolic and diastolic failure) |
| Sputum retention (postoperative, traumatic) |
| Pulmonary embolism |
| Pneumothoraces and bullae |
| Uncontrolled oxygen |
| Sedation |
| Medication – non-compliance or side-effects |
| Nutritional (K, PO4, Mg deficiency, CHO excess) |
| Sleep apnoea |
The most common bacterial isolates are Streptococcus pneumoniae and Haemophilus influenzae in 80% of exacerbations.9S. viridans,10Moraxella (previously Branhamella) catarrhalis,11Mycoplasma pneumoniae12 and Pseudomonas aeruginosa may also be found. Viruses can be isolated in 20–30% of exacerbations13 and include rhinovirus, influenza and parainfluenza viruses, coronaviruses and occasionally adenovirus, and respiratory syncytial virus. Whether these organisms are pathogens or colonisers is often unclear.
PNEUMONIA
Pneumonia has been estimated to account for 20% of presentations requiring mechanical ventilation.13 It is most commonly caused by S. pneumoniae and H. influenzae but Mycoplasma, Legionella, enteric Gram-negatives and viruses are occasional causes.
LEFT VENTRICULAR FAILURE
Left ventricular (LV) systolic failure may result from coexisting ischaemic heart disease, fluid overload, tachyarrhythmias or biventricular failure secondary to cor pulmonale. LV diastolic failure occurs commonly and is precipitated by hypoxaemia, tachycardia,14 pericardial constraint due to intrinsic positive end-expiratory pressure (PEEPi) or right ventricular (RV) dilation. Increased work of breathing related to COPD will also increase by up to 10-fold the amount of blood flow to the respiratory pump muscles,15 thereby causing an increased demand upon the overall cardiac output. In patients with borderline cardiac status, this may precipitate heart failure. The components of right and LV failure can be accurately distinguished by Doppler echocardiography. Pulmonary congestion can be difficult to diagnose because of the abnormal breath sounds and chest X-ray appearance which are commonly present in COPD. In a recent publication, 51% of patients with acute exacerbation of COPD had echocardiographic evidence of left heart failure (systolic 11%, diastolic 32%, systolic and diastolic 7%).16
DIAGNOSIS AND ASSESSMENT
DIAGNOSIS
The clinical examination findings of COPD depend upon severity.
A chest X-ray will commonly show hyperinflated lung fields, as suggested by 10 ribs visible posteriorly, six ribs visible anteriorly or large airspace anterior to heart (> 1/3 of the length of the sternum), flattened diaphragms (best seen on lateral chest X-ray) and a paucity of lung markings. Pulmonary hypertension is manifest by enlarged proximal and attenuated distal vascular markings and by RV and atrial enlargement. Lung bullae may be evident.
A high-resolution computed tomographic (CT) scan of the chest (1–2-mm slices) can demonstrate characteristic appearance and regional distribution of emphysema. It can also assess for coexistent bronchiectasis, LV failure17 and pulmonary fibrosis. Such scans are less sensitive than standard chest CT scans (1-cm slice) for detecting pulmonary lesions (e.g. neoplasms). Nuclear ventilation perfusion scans can also provide a characteristic appearance of COPD.
DIFFERENTIAL DIAGNOSIS
Chronic heart failure (CHF) may be a differential diagnosis of COPD, or simply coexist, as both disorders are common in smokers.14,16 Orthopnoea and paroxysmal nocturnal dyspnoea are features which correlate with heart failure severity. A past history of myocardial ischaemia or atrial fibrillation should alert one to the possibility of heart failure. An echocardiogram and high-resolution CT (looking for shift in interstitial oedema with changes in posture from supine to prone)17 are sensitive markers of CHF.
NON-VENTILATORY MANAGEMENT
OXYGEN THERAPY
Oxygen given by low-flow intranasal cannulae or 24–35% Venturi mask should be titrated to achieve a saturation (SpO2) of 90 ± 2% as these levels will avoid significant increases in PaCO2 in the majority of COPD patients with ARF. Increases in PaCO2 are most common in patients with initial PaCO2 > 50 mmHg and pH < 7.35.18 If the rise in PaCO2 is excessive (> 10 mmHg or 1.33 kPa), then FiO2 should be reduced, titrating SpO2 to 2–3% below the previous value, and arterial blood gases should be repeated. If no PaCO2 rise occurs with oxygen therapy, then a higher SpO2 may be targeted with repeat blood gases.
Inadequate reversal of hypoxia (e.g. SpO2 < 85%) is suggestive of an additional problem such as pneumonia, pulmonary oedema or embolus, or a pneumothorax. Investigation of this should commence and a higher O2 delivery system should be used (see Chapter 24). Although high levels of O2 should be avoided, reversal of hypoxia is important and O2 should not be withheld in the presence of hypercapnia, or withdrawn if it worsens.
BRONCHODILATORS
Bronchodilators are routinely given in all acute exacerbations of COPD because a small reversible component of airflow obstruction is common, and bronchodilators improve mucociliary clearance of secretions.19 However, a large meta-analysis of 22 large randomised controlled long-term trials of ambulatory COPD patients involving either anticholinergics and/or β2 agonists (short- and long-acting) over 3–60 months indicates that anticholinergics are more favourable than placebo in terms of acute exacerbations, hospitalisations and respiratory deaths.20 There were no favourable advantages with β2-agonists compared with placebo for acute exacerbations or hospitalisations, and placebo was better than β2-agonists in terms of respiratory death.20
ANTICHOLINERGIC AGENTS
Anticholinergic agents, such as ipratropium bromide, have been shown to have a similar or greater bronchodilator action than β-agonists in COPD,1,21,22 and also to have fewer side-effects and no tachyphylaxis. Anticholinergic agents should be used routinely in COPD with ARF and many now believe them to be the agent of first choice.1 An ipratropium bromide nebule of 0.5 mg in 2 ml should be nebulised initially 2-hourly, then every 4–6 hours. Long-term use of ipratropium bromide has been shown to reduce the incidence of exacerbations23 and is therefore recommended for chronic use in ambulatory COPD. Long-acting anticholinergics (e.g. tiotropium) offer potential of once-daily dosing.




Full access? Get Clinical Tree


