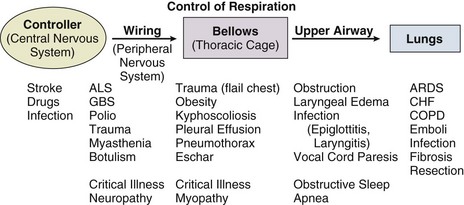
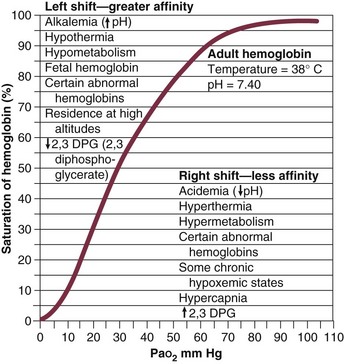
37 Acute respiratory failure is defined as the inability of the respiratory system to meet the oxygenation, ventilation, or metabolic requirements of the patient.1 Although the main function of the lungs appears to be related to gas exchange (i.e., oxygenation and ventilation), it should be remembered that the lung is a metabolically active organ as well.1,2 Respiratory failure has been divided into two main types. Type 1 is hypoxemic respiratory failure, and type 2 is hypercapnic failure with or without hypoxemic respiratory failure.2 More simply stated, type 1 respiratory failure is oxygenation failure and type 2 is ventilatory failure. Operationally, type 1 respiratory failure is defined by a partial pressure of oxygen in arterial blood (PaO2) less than 60 mm Hg and type 2 respiratory failure is defined by a partial pressure of carbon dioxide in arterial blood (PaCO2) greater than 50 mm Hg. The respiratory failure can be acute or chronic in nature, related to the onset and duration of the failure.2,3 Some patients may present with an acute deterioration or worsening of their chronic respiratory dysfunction termed acute-on-chronic respiratory failure.4 Acute respiratory failure is commonly encountered in the intensive care unit (ICU) setting and may be the primary reason for the admission or a complication of the patient’s medical condition(s) or treatment. Respiratory failure may be the result of a variety of causes, some of which may not directly involve the lungs or the respiratory muscles.2 Like a chain composed of individual links representing the brain, peripheral nervous system, upper airway, lower airway, respiratory muscles, cardiovascular system, and lungs (Fig. 37.1),2 respiratory failure may result when any of the links become sufficiently dysfunctional or weak. Like a chain, the body is only as strong as its weakest link, and respiratory failure may result when a component of the chain becomes sufficiently compromised. Common causes of hypoxemic and hypercapnic respiratory failure are listed in Box 37.1. This chapter reviews the basic mechanisms and clinical manifestations of type 1 and 2 respiratory failure and concludes with a more in-depth discussion of acute respiratory distress syndrome (ARDS), which includes clinical disorders in the type 1 category (ventilator management will be addressed in more depth in other chapters). Hypoxemic respiratory failure refers to the inability of the respiratory system to maintain satisfactory levels of oxygen in the arterial blood.3 The five basic mechanisms of hypoxemia are listed in Box 37.2. Ventilation-perfusion abnormality is the most common.2,3 Multiple mechanisms may be instrumental in the development of respiratory failure. Most of the abnormalities will improve with the administration of supplemental oxygen, except for a shunt abnormality in which the PaO2 continues to be low despite the administration of high levels of supplemental oxygen. The shunt may be either intracardiac (such as a right-to-left shunt through a patent foramen ovale) or intrapulmonary (as seen with pneumonia or ARDS).5,6 A diffusion abnormality is infrequently the cause of hypoxemia in clinical practice and typically is only significant in the setting of tachycardia, high cardiac outputs, and when the diffusion capacity is below 25% of predicted.3 The hallmark of type 1 respiratory failure is hypoxemia, which is primarily assessed by the PaO2 of an arterial blood gas (ABG). An estimate of the patient’s oxygenation status can be obtained by measuring the oxygen saturation using a pulse oximeter. An appreciation of the sigmoid shape of the oxyhemoglobin dissociation curve allows for a rough estimation of the PaO2 (Fig. 37.2). The curve will shift rightward or leftward in relation to changes in the pH, PaCO2, temperature, and PO4−2 concentration.7 Determination of the ABG and knowledge of the exact FIO2 allows for the calculation of the alveolar-arterial oxygen gradient3 (A-a gradient) using the formula PAO2 − PaO2 = A-a gradient; thus, where PAO2 Partial pressure of oxygen in the alveolus PaO2 Partial pressure of oxygen in the arterial blood FIO2 Fraction of inspired oxygen PH2O Water vapor pressure at standard temperature and pressure PaCO2 Partial pressure of carbon dioxide in the arterial blood The A-a gradient is FIO2 dependent and will increase as the FIO2 increases. Because of this FIO2 dependency, some have preferred to evaluate oxygenation abnormalities by calculating the PaO2/PAO2 ratio, which is independent of the FIO2.3 For simplicity’s sake, many individuals use the PaO2/FIO2 ratio as a measure of oxygenation abnormality. This value is used in the definition of ARDS and has gained wide-scale acceptance as a measure of abnormal oxygenation.8 The shunt fraction or venous admixture can also be calculated while a patient breathes 100% oxygen and has reached steady-state oxygenation. Hypercapnic respiratory failure can be the result of a variety of disorders as listed in Box 37.1.9 Acute hypercapnic respiratory failure, also termed acute ventilatory failure, occurs when a patient’s ABG reveals acute respiratory acidemia (pH <7.35) with a PaCO2 greater than 50 mm Hg. This definition is not generally applicable to patients with severe chronic obstructive lung disease or neuromuscular disorders who have developed a compensatory metabolic alkalemia in response to their chronic hypercapnia. However, these patients may have acute exacerbations or comorbid conditions that cause them to decompensate into acute-on-chronic respiratory failure. Not all respired air is effective alveolar ventilation because some of the total tidal ventilation (VT) ventilates nonperfused areas or dead space (VD) (both anatomic and pathologic). This relationship between tidal volume, dead space volume, and alveolar volume can be expressed as3: The minute ventilation (VE) is equal to the sum of the dead space ventilation (VD) and the alveolar ventilation (VA): VE = VD + VA. Combining and rearranging terms from these various equations yields the following relationship of carbon dioxide production and alveolar ventilation with the PaCO2.3 Therefore, elevated PaCO2 can result from a combination of any of three clinical alterations: increased CO2 production, decreased tidal ventilation, or increased dead space ventilation.9 Increased CO2 production arises from hypermetabolic states such as exercise, fever, sepsis, burns, trauma, excessive carbohydrate intake, and hyperthyroidism.9 In isolation, these relatively common conditions rarely induce hypercapnic respiratory failure because the normal physiologic response to an elevated PaCO2 is to increase minute (and therefore alveolar) ventilation to maintain eucapnia and a normal pH. Only those patients who are unable to increase their effective alveolar ventilation as a result of neuromuscular disorders affecting the muscles of respiration, the presence of excessive ventilation/perfusion ( Decreased tidal volume hypoventilation can be caused by disorders affecting any component of the “neuromuscular sequence” starting in the central nervous system (CNS) and ending at the muscles of respiration (see Fig. 37.1). Among the more common causes are CNS depressants such as narcotics and sedatives, which diminish respiratory drive; cerebral vascular disorders, especially those that involve the brainstem, which impair the efferent signals to breathe; and disorders of neuromuscular transmission such as myasthenia gravis and the paraneoplastic Eaton-Lambert myasthenic syndrome (Box 37.3).10,11 Abnormal respiratory mechanics can result from airflow obstruction, chest wall deformities, and loss of lung volume. Disorders that increase the respiratory load, such as a circumferential chest burn eschar or flail chest, or impair function of the respiratory muscles, such as kyphoscoliosis, will also lead to hypoventilation. The hallmark feature of inadequate ventilation is the presence of an elevated PaCO2 with or without the presence of hypoxemia.3,9 In the adult population the best method to detect an elevated PaCO2 is with an ABG measurement. Noninvasive evaluation of ventilation in the critically ill adult, using end-tidal CO2 (PetCO2) measurements or transcutaneous CO2 (PtcCO2) determinations, is not as reliable as it is in the healthy adult or the neonate, respectively.7 The risk factors for developing acute respiratory failure include the postoperative state, preexisting chronic illness, malnutrition, advanced age, morbid obesity, chronic bronchitis, and cigarette smoking. The clinical manifestations of respiratory distress may be subtle and nonspecific or may be obvious to even the untrained observer. The patient with respiratory distress may have an abnormal respiratory rate (too high or low) or display an irregular pattern of breathing. The patient with respiratory distress may evidence gasping ventilation, nasal flaring, or use of the accessory muscles of respiration. Intercostal retractions may be seen, and the patient may exhibit paradoxical respiratory movement. Typically, patients with acute respiratory failure, whether type 1 or type 2, will have altered heart rate and blood pressure. The majority will evidence a sympathetic response with tachycardia and hypertension, but patients may be hypotensive and bradycardic. Cardiac arrhythmias may be seen in both type 1 and type 2 respiratory failure. Patients with acute respiratory failure will usually be in distress and often appear apprehensive.2,9 Many will be diaphoretic and have altered mental status or level of consciousness. Hypercapnic patients may demonstrate signs of a respiratory encephalopathy including somnolence, coma, asterixis, seizures, tremors, or myoclonic jerks. Papilledema and congested conjunctivae may be present. Type 1 respiratory failure patients may appear cyanotic.9 Since the initial 1967 description of acute catastrophic respiratory failure in 12 patients who were subsequently declared to have the acute respiratory distress syndrome, there has been a great deal of research and clinical study attempting to understand the mechanism of injury and improve the outcome of these critically ill patients.12–14 The hallmark of this disorder was the rapid onset of acute hypoxemic respiratory failure that was characterized by refractory hypoxemia despite the administration of high concentrations of supplemental oxygen; decreased pulmonary compliance; diffuse, bilateral pulmonary infiltrates; the absence of left-sided heart failure; and histologic evidence of alveolar damage with hyaline membrane formation, which was followed by fibrosis of the lung.8,12,13 The subsequent 10 to 20 years of ARDS management were associated with extremely high mortality rates, at times approaching 90%, despite the provision of extremely aggressive supportive care.15–20 Some of the initial clinical and basic investigations that attempted to improve the outcome of patients with ARDS were hampered by the lack of uniformly accepted definitions.21–25 In 1994 an American-European Consensus Conference (AECC) defined acute lung injury (ALI) and ARDS in an attempt to eliminate confusion related to terminology for these conditions.21,22 The definitions are clinically based and regard ALI as a continuum with the more severe oxygenation abnormalities reflective of ARDS. Among the main benefits of the uniform definition would be improved communication and enhanced clinical trial design. The AECC definition of ALI and ARDS was challenged and there were noted discrepancies between the clinical and pathologic findings in individuals felt to have died with ARDS.25 Esteban and colleagues25 found that one third of the people who died with a clinical diagnosis of ARDS did not have histologic evidence of diffuse alveolar damage on postmortem examination. In addition, diffuse alveolar damage, the hallmark pathologic finding of ARDS, was evident in 10% of patients who died without a clinical diagnosis of ARDS. A subsequent study evaluated the sensitivity and specificity of the Murray Lung Injury Score, the AECC definition of ARDS, and a Delphi definition that incorporated oxygenation abnormality, X-ray findings, clinical onset, compatible clinical scenario, and absence of left-sided heart failure for ARDS.26 The combination of Lung Injury Score greater than 2.5 with either the AECC definition or the Delphi definition of ARDS resulted in the best combination of sensitivity and specificity. Since its introduction, the AECC definition has been used by the vast majority of clinical trials evaluating new treatment strategies for patients with ARDS, particularly those conducted by the ARDS Network investigators, supported by the U.S. National Heart, Lung, and Blood Institute of the National Institutes of Health.27–29 In 2011 experts from the European Society of Intensive Care Medicine, the American Thoracic Society, and the Society of Critical Care Medicine met in Berlin and proposed a new definition for staging ARDS as mild, moderate, or severe, based on the oxygenation impairment measured on a minimum of 5 cm H2O positive end-expiratory pressure (PEEP) (10 cm H2O for severe).30 The new Berlin definition did not drastically differ from the prior AECC definition, but it clarified several important concepts: 1. Timing—patients with ARDS should be identified within 72 hours of a recognized risk factor and nearly all patients should be identified within 7 days. 2. Chest imaging—bilateral opacities consistent with pulmonary edema are seen on chest radiograph or chest computed tomography (CT) scan. 3. Origin of pulmonary edema—ARDS may be present in the setting of coexisting cardiogenic edema or volume overload if in the opinion of the treating physicians the respiratory failure is not fully explained by these conditions and there is a recognized risk factor for ARDS. In the absence of a recognized risk factor for ARDS there should be some objective assessment of cardiac function such as echocardiography or pulmonary capillary wedge pressure measurement. 4. Oxygenation—because PEEP can affect the PaO2/FIO2 ratio there should be a minimum of 5 cm H2O PEEP and 10 cm H2O in those patients with severe ARDS (PaO2/FIO2 < 100).30 The Berlin definition for ARDS eliminates the use of the term “acute lung injury” to classify the severity of lung injury and defines mild ARDS as a PaO2/FIO2 300 or less but greater than 200, moderate ARDS as a PaO2/FIO2 200 or less but greater than 100, and severe ARDS as a PaO2/FIO2 100 or less, with morality rates associated with the different stages 27%, 32%, and 45%, respectively.30 To date there is no specific laboratory test that is pathognomonic for the diagnosis of ARDS.8,21,27 All of the current definitions for ARDS require the presence of bilateral pulmonary infiltrates on chest imaging, but interpretation is not always straightforward.21,31 When 21 experts were given 28 chest radiographs to evaluate for the presence of bilateral pulmonary infiltrates, they only agreed 43% of the time, and one third of the time five or more individuals differed in their interpretation.29 The range of ALI/ARDS diagnoses was from 36% to 71%.32 It has been recognized that ARDS may arise in association with a number of clinical conditions or risk factors, with sepsis being the most common (Box 37.4).8,18,21,27,33–37 Various studies report that approximately 5% to 40% of septic patients will develop ARDS.8,18,36 Shock (prolonged hypotension) and the systemic inflammatory response syndrome (SIRS) are also common risk factors. Other frequently encountered clinical risk factors include multiple emergency transfusions; aspiration injury; near-drowning; pancreatitis; trauma (particularly lung contusion, fat emboli from long bone fractures); burns; cardiopulmonary bypass; and disseminated intravascular coagulation (DIC). These clinical risk factors are synergistic, and when more than one of the clinical risk factors is present, the likelihood of ARDS is greater than just the sum of the collective risk factors.8,17 The clinical risk factors associated with the development of ARDS appear to greatly influence the expected outcome. Whether ALI and ARDS arise from a direct pulmonary insult or from an extrapulmonary cause impacts the subsequent response to PEEP and ventilatory support (Box 37.5).19 Age also has an impact on the likelihood for ARDS development.38 Older individuals have a greater likelihood of developing ARDS from most of the risk factors noted earlier. In the setting of trauma the risk for ARDS seems to peak in the sixth and seventh decades and then declines.38 Negative pressure (postobstructive) pulmonary edema (NPPE or POPE) is a type of noncardiogenic pulmonary edema that can manifest as ARDS.39 It is rare (five cases reported from a large tertiary medical ICU in 4 years),40 can range in severity from mild hypoxemia treated with low-flow supplemental oxygen to profound hypoxemia requiring mechanical ventilation, and may even present as alveolar hemorrhage.41–45 It is believed to be caused by increased vascular permeability resulting in alveolar and capillary damage from large levels of negative intrathoracic pressure generated by attempting to inspire against an occluded airway or airway obstruction.46,47 This leads to a protein-rich alveolar exudate impairing gas exchange in mild-to-moderate cases and leaking of capillary blood into the alveoli in more severe circumstances. Most of the reported cases are related to surgical procedures: thyroidectomy,48,49 septorhinoplasty,50 tonsillectomy/adenoidectomy,51 mandibular open reduction and internal fixation,52 or cryosurgery for a tracheal obstruction.53 Nonoperative conditions leading to NPPE have included laryngospasm,54,55 occlusion of an endotracheal tube,56,57 excessive tube thoracostomy suction pressure,58 nonlethal hanging,59 and even hiccups.60,61 The common element is the rapid relief of a large airway occlusion. Patients often have no underlying cardiopulmonary disorders. NPPE may account for a small percentage of immediate postoperative extubation failures, particularly if an unrecognized mainstem intubation is present in an awake and spontaneously breathing patient. Clinical manifestations of hypoxemia and respiratory distress may be delayed up to 6 hours.47 Therapy is supportive with supplemental oxygen and, if needed, positive-pressure ventilation (invasive or noninvasive). Elevated intracranial pressures, either from traumatic brain injury or intracerebral hemorrhage, or status epilepticus can induce an ARDS-like clinical syndrome termed neurogenic pulmonary edema (NPE).62,63 Although the exact mechanisms are not clear, an abrupt and massive release of catecholamines from injured neuronal tissues is thought to act directly on the lung parenchymal vasculature to promote a “capillary leak” of a protein-rich exudate as well as an increase in pulmonary vascular resistance64 in addition to inducing a neurohumoral (takotsubo) cardiomyopathy.65–67 The cardiomyopathy has the distinctive echocardiographic feature of ventricular apical ballooning and can persist for months.68 The exact incidence and prevalence of ARDS has been variable and related to definitions used (Box 37.6).8,38 Early reports suggested that there were 150,000 patients with ARDS each year in the United States.8,33,36,69 This statistic led to an estimated incidence of 75 cases per 100,000 population. Proposed estimates have suggested that the incidence of ARDS ranges from 1.5 to 64 cases per 100,000 population.38,69,70 A recent study evaluated the incidence and outcome of ALI in mechanically ventilated patients aged 15 or older cared for at 21 hospitals in and around King County, Washington, over a 15-month observation period.38 The crude incidence of ALI was 78.9 per 100,000 person-years, and the incidence of ARDS was 58.7 per 100,000 person-years. On the basis of their results, the authors estimated the annual number of U.S. episodes was 332,100 cases of ARDS. The authors noted that increasing age was associated with an increased incidence and mortality rate in ARDS. A recent prospective observational study using the AECC definition and lung protective ventilatory support strategies in Spain (the ALIEN study) reported an incidence of 7.2 per 100,000 population per year for ARDS with an ICU and hospital mortality rate of 42.7% and 47.8%, respectively.71 When ARDS becomes clinically apparent, the patient is usually noted to be in significant distress manifesting dyspnea, tachypnea, visible signs of respiratory distress, and increased work of breathing.8,23,33,36 The typical presentation is manifest as an acute catastrophic complication in a patient who has one or more of the clinical risk factors for the development of this form of ARDS. The precipitating injury need not directly involve the pulmonary system.8,36,72 Past definitions have emphasized the need to exclude patients with previous or known chronic pulmonary or cardiovascular diseases.19–21,27 The AECC definition excluded patients with elevated left-sided heart filling pressures and chronic infiltrative lung disease as the cause of the radiographic or physiologic alterations, but the new Berlin definition will allow for the concomitant presence of ARDS and cardiac edema/volume overload as long as the predominant process is thought to be ARDS.21,30 The hallmark of ARDS is the presence of hypoxemia despite the administration of high concentrations of inspired oxygen, evidence of an increase in the shunt fraction, a decrease in pulmonary compliance, and an increase in the dead space ventilation.8,24,30 The chest radiographic manifestation of ARDS is the presence of diffuse, bilateral pulmonary infiltrates with a normal cardiac silhouette. Recent reports have cautioned that even among trained experts there is often disagreement concerning the interpretation of the chest radiograph.31,29,69,73 Chest CT has also demonstrated that the radiographic injury is not homogeneous and has a predominance in the dependent portions of the lung.8,43 It is important to remember that ARDS is a clinical syndrome and the diagnosis is made clinically, not on the basis of a single radiograph, ABG analysis, or laboratory test. Typically, type 1 alveolar cells compose the major gas exchange surface of the alveolus and are integral to the maintenance of the permeability barrier function of the alveolar membrane.8 Type 2 pneumocytes are the progenitors of type 1 cells and are responsible for surfactant production and homeostasis.8 During ARDS there is damage to the capillary endothelial and the alveolar epithelial cells.8 Cellular injury and alteration of the normal barrier function results in a permeability defect that gives way to flooding of the alveoli with protein-rich fluid and inflammatory cells.8,33,74 This results in the alteration of pulmonary mechanics, physiology, and gas exchange.8,74 There is alteration of surfactant that results from damage to the type 2 pneumocyte and from the inactivation and dilution of alveolar surfactant from the protein and fluid that have entered into the alveolar space, respectively.75,76 Surfactant dysfunction can lead to atelectasis and a further reduction in pulmonary compliance.8,75–78 In addition, dysfunction of the alveolar epithelial cells can impair the resorption of fluid from the alveolar space, which augments the parenchymal injury process and gas exchange abnormalities.8,79 The observed pathologic findings in ARDS depend on the timing of the tissue sampling. During the initial stages of clinically evident lung injury there is histologic evidence of diffuse alveolar damage.8 Histologic features of the injury include microthrombi composed of platelets and white blood cells within the capillary lumen, denudation of the alveolar epithelial lining cells, swelling of the capillary endothelial cells, interstitial and alveolar infiltration by polymorphonuclear leukocytes (PMNLs), and hyaline membrane formation within the alveoli.8 Grossly, the lungs appear heavy and wet. Later, areas of type 3 collagen deposition with fibrosis will be present.79 An intense inflammatory reaction involving PMNLs, activated monocytes, macrophages, and endothelial cells is present in the fibroproliferative phase of lung injury.76,80,81 Pro- and anti-inflammatory molecules produced by these activated cells may be found in the circulating blood and bronchoalveolar lavage (BAL) fluid.81–83 This phase may be followed by fibrosis, but this fibrosis does not appear to have the same permanence as typical fibrosis would have and may actually resolve over time in survivors of the injury.84 ARDS may develop as a result of epithelial or endothelial cell injury.8,13,85,86 Both sites of injury and cells are important for maintenance of normal barrier function and are capable of initiating an inflammatory response. In the majority of clinical settings the initial site of the ARDS involves the capillary endothelial cell, which may be the initial manifestation of a “panendothelial cell injury” resulting from SIRS.83 Endothelial cell injury compromises the integrity of the vascular barrier and results in transudation of fluid and inflammatory mediators into the interstitial tissues and ultimately into the alveoli.8,13 The frequent occurrence and early involvement of lung dysfunction as a component of multiple organ dysfunction/failure lends support to the hypothesis of a panendothelial cell injury as one of the target injuries in the setting of SIRS.87 The complex pathophysiologic processes that culminate in the production of ARDS involve a delicate balance between the body’s proinflammatory and anti-inflammatory responses to the inciting clinical event.83,87 The balance that exists between the various inflammatory molecules or mediators and the endogenous compensatory responses evoked by the inflammatory response will dictate whether or not lung injury and other forms of organ dysfunction will develop.81 The ensuing interaction between SIRS and the compensatory anti-inflammatory response syndrome (CARS) will thus determine whether a patient successfully deals with an injury or is predisposed to develop organ dysfunction (excessive SIRS response) or immunosuppression and infectious complications (excessive CARS response).83,87 This mixed antagonistic response syndrome (MARS) has tremendous impact on the fate of the critically ill patient.87 Basic management strategies for patients with ARDS are listed in Box 37.7.8,18,21,23,33,36,88 Initial attention must be directed to providing lung protective ventilator support and identification of the predisposing underlying clinical risk factor(s) or condition(s), and there should be specific treatment directed at the underlying or predisposing disorder.8,21–23 The recognition of sepsis and infection as frequent causes of ARDS should prompt an aggressive search for undiagnosed foci of infection and the administration of appropriate antimicrobial treatment and use of surgical drainage procedures as indicated.15,17,89 The cornerstone of supportive management is the provision of mechanical ventilatory support.8,21–23 Recent experimental and clinical data report significant survival benefit from the use of lung protective ventilatory support strategies.90,91 The primary goal of this support is to improve oxygenation and ensure that the lung is allowed to heal and avoid further injury. Use of nonprotective ventilatory support strategies may have a role in the persistent proinflammatory response and the development of multiple organ dysfunction syndrome (MODS) and multiple organ failure (MOF).90–93 See Chapter 11 for a more detailed discussion of mechanical ventilatory support in the management of patients with ARDS. Over the past 20 years there has been increasing recognition that the ventilatory support strategy used in the management of patients with ARDS may produce or augment lung injury or impair the healing process (Box 37.8).94–98 Results of experimental animal studies demonstrated clinical, physiologic, and histologic abnormalities similar to those observed in patients with ARDS when animals with normal lungs were ventilated with large tidal volumes or were given high inflation pressures.95–101 The alveolar overdistention produced by these ventilatory modes was felt to be the critical element in production of the lung injury.95–98 Lung injury could result from the administration of large tidal volumes or the administration of positive pressure or negative pressure breaths that were sufficient to produce alveolar overdistention (termed volutrauma).97–102 Other mechanisms that could potentially result in lung injury were the repetitive recruitment-derecruitment of distal airways (termed atelectotrauma) and alveoli or the disruption of alveoli resulting in translocation of organisms or air emboli.97,98 Alveolar overdistention can also give rise to systemic inflammatory molecules that may contribute to the SIRS response and drive the development of MODS/MOF.92,98 This has been termed biotrauma.98 Interestingly, the lung injury produced by high inflation pressures or large tidal volumes in these experimental models could be ameliorated by the addition of therapeutic amounts of PEEP.97,98 Subsequent human studies have evaluated the distribution of the radiographic lung injury in ARDS patients as determined by the use of CT scans of the chest and have noted the dependent nature of the injury.74,103 The injury to the lungs was not diffuse and homogeneous as once believed. These dependent areas of injury comprised regions of alveolar flooding from the gravitational accumulation of lung water, areas of lung injury, and normal lung regions. The relatively normal ventral (nondependent) lung has been referred to as the “baby lung,” and the injured (dependent) lung has been called the “sponge lung.”32,103 With the use of higher tidal volumes, PEEP, or high distending pressures, there was evidence of alveolar overdistention, particularly in the areas of normal lung. A multicenter trial of low versus traditional tidal volume ventilation in patients with ARDS demonstrated increased proinflammatory cytokines in the serum and BAL fluid of patients ventilated with the larger traditional tidal volumes, supporting the concept of biotrauma from alveolar overdistention.92
Acute Respiratory Failure
Acute Respiratory Failure—Types 1 and 2
Hypoxemic Respiratory Failure
Basic Mechanisms
Assessment of Oxygenation
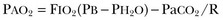
Hypercapnic Respiratory Failure
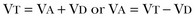
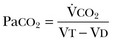
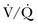 ) mismatching, or increased dead space ventilation develop hypercapnic respiratory failure caused by elevated CO2 production.
) mismatching, or increased dead space ventilation develop hypercapnic respiratory failure caused by elevated CO2 production.
Assessment of Ventilation
Clinical Manifestations
Acute Respiratory Distress Syndrome
Risk Factors
Incidence and Prevalence
Clinical Manifestations
Pathologic Manifestations
Pathophysiology
Management Strategies
Mechanical Ventilation
Related posts:

Full access? Get Clinical Tree


Acute Respiratory Failure
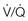 mismatch
mismatch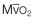
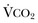 ) equals the rate of CO2 elimination. Carbon dioxide elimination (
) equals the rate of CO2 elimination. Carbon dioxide elimination (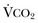 ) is equal to the alveolar ventilation (V
) is equal to the alveolar ventilation (V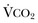 = V
= V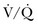 abnormalities, to account for the additional hypoxemia in these patients with hypercapnic respiratory failure.
abnormalities, to account for the additional hypoxemia in these patients with hypercapnic respiratory failure.




