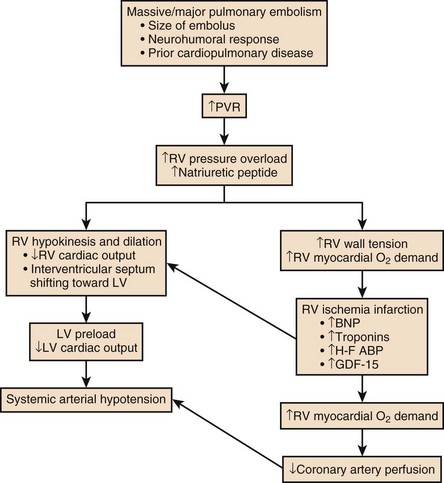
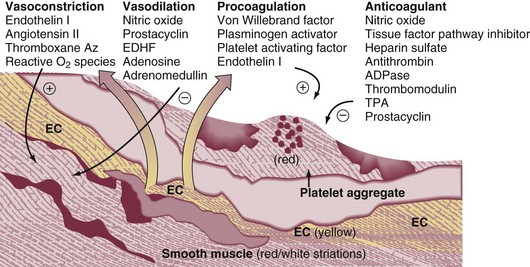
44 PREVALENCE OF VENOUS THROMBOEMBOLISM IN INTENSIVE CARE UNIT PATIENTS Clinical Presentation on Admission to the Intensive Care Unit Recognition of Pulmonary Embolism During Intensive Care Unit Admission DIAGNOSTIC TESTING FOR PULMONARY EMBOLISM Arterial Blood Gas Measurement Duplex Compression Ultrasonography Ventilation-Perfusion Nuclear Scans Multidetector Computed Tomography Scan of the Chest THE MANAGEMENT OF ACUTE PULMONARY EMBOLISM Pulmonary venous thromboembolism (VTE) and deep venous thrombosis (DVT) are different manifestations of the same disease. Despite adequate VTE prophylaxis in the intensive care unit (ICU) the presence of a VTE event is a constant threat to critically ill patients. Although the critical care clinician may encounter the embolization of air, fat, infected clots, amniotic fluid, tumor, and inorganic substances, by far VTE will be the most common pulmonary embolic condition encountered. Critical care clinicians will encounter life-threatening pulmonary embolism (PE) ranging from hemodynamically stable patients with varying degress of right ventricular dysfunction (RVD) to acute massive PE, defined as hemodynamic shock from acute PE, which represents the most serious manifestation along the spectrum of venous thromboembolic disease.1–3 According to population-based studies, the annual incidence of VTE in the United States has ranged from 200,000 to a recent estimate of 900,000 cases in which approximately 300,000 people die every year from acute PE.4,5 More recently, in 2006, 467,000 patients were hospitalized with DVT and 247,000 patients were admitted with acute PE.6 VTE is associated with a significant health care costs. Based on 2004 provider payments, the economic burden of VTE costs ranges between $5.8 billion and $7.8 billion.7 The mortality rate can exceed 58% in patients with acute PE presenting with shock, and most of these deaths occur within 1 hour of presentation.1,2 Acute PE is the third most common cause of death among hospitalized patients, and with an aging population the number of people with VTE is expected to increase. For these reasons, the U.S. Surgeon General issued a “Call to Action” in 2008, identifying VTE as a major public health problem.8 The development of DVT occurs largely in the lower extremities and can result in the sequelae of PE, post-thrombotic syndrome, and chronic thrombotic pulmonary hypertension.9 Venous thrombosis is associated with significant morbidity and mortality rates but can be prevented in most patients.2,3 ICU patients represent a heterogeneous population. In prospective screening studies for DVT, in the absence of thromboprophylaxis, the incidence of DVT in a medical-surgical ICU ranges from approximately 13% to 33%.10–12 In an autopsy review of six studies of 436 critically ill patients the incidence of PE ranged from 7% to 27% (mean 13%); PE contributed or directly caused death in 0% to 12% (mean 3%) of the patients. It is important to note that in a majority of the patients there was no antemortem suspicion of fatal PE.13 Contemporary prevalence of VTE that are admitted to or occur in the ICU setting can be obtained from large VTE clinical trials. Approximately 10% of patients entered into the PIOPED-I (Prospective Investigation of Pulmonary Embolism Diagnosis) study for possible PE were hospitalized in a medical or surgical critical care unit.14 In the recent international, multicenter EINSTEIN PE trial,15 which randomized 4823 symptomatic PE patients to rivaroxiban or enoxaparin and a vitamin K antagonist, 600 patients (12.4%) required ICU admission. Finally, in the PROTECT trial (Prophylaxis for Thromboembolism in Critical Care Trial)16 among 3764 critically ill patients enrolled in which a majority were on mechanical ventilator support and 40% were on vasopressor therapy, dalteparin and unfractionated heparin (UFH) were the agents used for VTE prophylaxis. The primary end point was proximal DVT presence. Using compression ultrasonography (CUS), 3.5% of the patients had proximal DVT on admission. The incidence of proximal DVT was 5.1% in the dalteparin group and 5.4% in the UFH group. The proportion of patients with pulmonary emboli was significantly lower with dalteparin (24 patients, 1.3%) than with UFH (43 patients, 2.3%).16 There is controversy over routine DVT screening in the ICU. As mentioned earlier, most ICU patients with DVT are asymptomatic and the clinical signs for DVT are nonspecific. CUS is time consuming and not cost effective. Efforts must be made to ensure adherence to current DVT prophylaxis guidelines. In ICU patients in whom these guidelines cannot not be fully implemented (intracranial, spinal, or leg injuries, or patients at increased bleeding risk) then routine CUS screening may be appropriate.17–21 In the nineteenth century Virchow described stasis, vascular wall abnormalities, and hypercoagulability as general risk factors for the development of DVT.22 As illustrated in Table 44.1, patients admitted to the ICU possess multiple preexisting VTE risks factors on admission to the ICU, yet in the International Cooperative Pulmonary Embolism Registry (ICOPER) 20% of the patients had idiopathic or unprovoked PE.3 The interaction between patient-related and setting-related risk factors appears to be responsible for the development of the VTE.23–25 Patient-related risk factors are usually acquired and are longstanding. Examples of patient-related risk factors are age, history of prior VTE, malignancy, neurologic and medical illnesses that result in immobility, cardiopulmonary disorders, collagen vascular disease, and vasculitis. In addition, patient-related factors may include genetic and acquired thrombophilia and hormone replacement therapy and oral contraceptive therapy. Setting-related risk factors are usually temporary. Examples of setting-related risk factors include hospitalizations, nursing home placement, and admission to the ICU. It is estimated that as high as 95% of clinically significant pulmonary emboli originate from the deep veins of the lower extremity. The proximal deep veins of the leg are the most common sites of origin of clots that embolize to the pulmonary circulation. A general consensus suggests that clinically significant proximal DVTs and risk for PEs originate by proximal extension of calf DVT.47,48 Venous thrombi are created in the setting of low flow and low shear stress. These thrombi consist of fibrin strands, red blood cells, and platelets. The thrombi form in the valve pockets of calf veins and extend to the proximal veins.48 After the thrombosis is formed there is raised venous capillary pressure, which increases the transcapillary filtration and the pressure rate, resulting in edema. In approximately 50% of patients, venous outflow obstruction decreases within 3 months by lysis and recanalization.49 Patients who have early edema are most likely to have residual thrombosis. Clots that continue to propagate have a greater risk to break apart and lead to embolization. This generally occurs more frequently in the first few days after clot formation. Clots that do not continue to propagate resolve by either fibrinolysis or organization. These processes generally occur within 7 days of clot formation.49,50 In general only 25% of patients with suspected DVT actually have it.47 The detection of DVT in critically ill patients is hampered by the patient’s inability to report symptoms and the unreliability of physical signs of DVT. CUS is noninvasive and highly sensitive and specific for the diagnosis of proximal lower extremity DVT.51 Full compressibility of either the femoral or popliteal veins excludes proximal DVT51 when compared to the gold standard demography; CUS has a sensitivity of 97% to 100% and a specificity of 98% to 99% for the detection of proximal thrombosis. Ultrasonography is less accurate in the diagnosis of distal calf vein thrombosis.51,52 Whether high-risk critically ill patients should be systematically screened for DVT using CUS, given the high incidence of DVT and limitation of clinical examination, is unresolved.13,17 Upper extremity deep venous thrombosis (UEDVT) accounts for approximately 4% of the venous thromboembolic events.53 Primary UEDVT makes up one third of all UEDVTs. Primary UEDVTs appear to be associated with inherited thrombophilic conditions, and the risk markedly increased to 14-fold when oral contraceptives were used.54 Catheter UEDVTs are more associated with the placement of a central venous catheter, and these patients are more likely to be inpatients than outpatients, as demonstrated by a large registry.55 In addition, patients with cancer have an increased occurrence of UEDVT when a central venous catheter is placed in these areas.55 Catheter-related UEDVT may have a higher pulmonary embolic potential than primary UEDVT.52 Nearly two thirds of patients who die from pulmonary thromboembolism die within 1 hour of presentation, but anatomically massive pulmonary emboli (>50% obstruction of the pulmonary circulation) are responsible for only half of the deaths. The term major pulmonary thromboembolism has been used to describe any pulmonary thromboembolus that results in a hemodynamically significant event.1 In patients with pulmonary thromboembolism, hemodynamic presentation is an important predictor of survival. The Urokinase Pulmonary Embolism Trial (UPET) demonstrated that the presence of hemodynamic decompensation was associated with a sevenfold increase in mortality rate.56 ICOPER confirmed these results by demonstrating a fourfold increase in mortality rate for those patients with hemodynamic instability.3 Because of the associations between outcome from pulmonary thromboembolism and shock or hypotension, aggressive intervention in patients thought to have pulmonary thromboembolism has given rise to the term golden hour, during which timely diagnosis and treatment are paramount.1 The principal pathophysiologic effects of pulmonary thromboembolism result from the acute impaction of material into pulmonary circulation and the resulting vascular obstruction and humoral mediator release as depicted in Figure 44.1.57 In nonthrombotic obstruction of the pulmonary vasculature, obliteration of 60% to 70% of the vascular tree is required to cause an elevation of the pulmonary artery pressure. In contrast, only 30% of the vascular tree must be obstructed in pulmonary thromboembolism for elevation of the pulmonary artery pressure to be achieved.58,59 Therefore, factors other than simple mechanical obstruction of the pulmonary vascular system play a role in the elevation of the pressures in the pulmonary vasculature during pulmonary thromboembolism. The production of platelet-derived vasoconstrictors may play a role in augmenting the increase in pulmonary vascular resistance (PVR) (Fig. 44.2).57,58,60 Thromboxane A2 (TxA2) is a potent vasoconstrictor and is the end product of arachidonic acid metabolism. TxA2 is produced by endothelial cells, and even in greater quantities by platelets in response to platelet aggregation. It has been demonstrated that there is increased production of TxA2 in the early phase of PE, which contributes to the PVR elevation and may be attenuated by cyclooxygenase (COX) inhibition in animal models.57 Serotonin (5-hydroxytryptamine) is one of the most potent pulmonary vasoconstrictors. Infusion of serotonin in an animal model can actually simulate the signs and symptoms of PE. Like TxA2, platelets are the primary source of serotonin production in PE. Serotonin antagonism can reduce PVR.57,60 There appears to be nearly an exponential increase in VTE events with age.29 The elderly demonstrate an incidence of 300 to 500 cases/100,000 persons compared to 30 cases/100,000 of persons age 25 to 35 years.29 A longitudinal study of 855 men followed from the ages of 50 to 80 years demonstrated the cumulative incidence of a VTE event was 0.5% by the age of 50 years and increased to 10.7% by the age of 80 years.3 The risk of VTE recurrences increases with age. In the elderly the PE rates are rising faster than the DVT rates; thus, a VTE event in an elderly person is more likely to be a PE, with its potential lethality.3,30 Anderson and Spencer31 reviewed multiple risk factors associated with a diagnosis of VTE and found that the risk factors were not equal in their impact on VTE occurrence. The strongest risk factors were hip and leg fractures, major general surgery, hip or knee replacement, major trauma, and spinal cord injury. Moderate risk factors included arthroscopic knee surgery, central venous lines, chemotherapy, congestive heart failure, hormonal replacement or oral contraceptives, cancer, pregnancy, thrombophilia, and prior VTE.31 Heit and associates32 found that the hospital setting, nursing home, or other chronic facility confinement was an independent risk factor for the VTE. This is most likely reflective of the various degrees of immobility that these patients experience. Several acquired and inherited hypercoagulable states have been shown to increase the risk of VTE and are collectively known as thrombophilia. Major acquired hypercoagulable conditions include malignancy, myeloproliferative disorders, and antiphospholipid syndrome (APS).33 DVT, the most common manifestation of the APS, occurs in 29% to 55% of patients with the syndrome, and about half of these patients have pulmonary emboli.34–36 The occurrence of a VTE event in a young adult alerts the clinician to the potential of an underlying inheritable thrombophilic condition. Nearly half the patients with a VTE event before the age of 45 have an associated inherited disorder.33 Factor V Leiden mutation causing resistance to activated protein C is the most common inherited risk factor. Factor V Leiden mutation is present in up to 5% of the normal population and is the most common cause of familial thromboembolism.33 Factor V Leiden mutation appears to be more associated with the development of DVT rather than PE.37 Primary or acquired deficiencies in protein C, protein S, and antithrombin III are other risk factors and their thrombotic expression is variable.33 Several primary disorders are associated with a high risk for fatal VTE. Patients frequently present with massive PE, and ICU admission and management is required. From the RIETE registry, risk factors of immobilization secondary to neurologic disease, cancer, and cardiopulmonary disorders had two- to threefold increased risk for fatal PE.38 In hospitalized cancer patients, VTE is the second leading cause of death.39 The risk of VTE in cancer patients undergoing surgery is three to five times greater than that in surgical patients without cancer. Moreover, cancer patients with symptomatic DVT exhibit a high risk of recurrent VTE.40 Heart failure is a frequent admission diagnosis in the coronary care unit. In patients with severe heart failure (ejection fractions less than 30%) PE is a major cause of morbidity and mortality rates.41,42 Among 198 patients with severe heart failure in the coronary care unit, 9.1% developed PE during their hospitalization, despite a majority receiving thromboprophylaxis.41 In an autopsy series of cardiac patients nearly a quarter had PE and the diagnosis was unsuspected in 82% of the patients.43 Acute exacerbation of chronic obstructive pulmonary disease (COPD) is a common reason for admission to the general hospital ward service and the ICU. Increase work of breathing leads to immobility, which is a risk factor of DVT. In a prospective cohort of 196 patients with COPD exacerbation, all patients had ultrasonography of the lower extremities performed. Among the 196 patients, 21 (10.7%) had DVT and the majority was asymptomatic.44 Moreover, hospitalized patients with unexplained exacerbations of COPD, when routinely evaluated, show demonstrated PE in 25% to 29%.45,46 As shown in Figure 44.1, the degree of mechanical obstruction, the interplay of neurohumoral mediators, and the patient’s underlying cardiopulmonary status will dictate the degree of RVD. A rapid rise in the pulmonary artery resistance produces a significant rise in right ventricular (RV) afterload, thereby causing RV dilatation. The dilation of the right ventricle results in shifting of the interventricular septum into the left ventricle, decreasing left ventricular (LV) preload.61–63 Moreover, RV dilation potentially promotes an increase in constrictive forces by the pericardium, leading to reduced LV compliance. Decreasing LV preload and compliance subsequently lead to lowering of the cardiac output and systemic hypotension with resultant decrease in aortic perfusion pressure and decreasing right coronary perfusion pressure.64,65 A compromised and burdened right ventricle is a direct result of oxygen demand outstripping oxygen supply, creating an ischemic environment for the right ventricle. The functional status of the cardiopulmonary system is very important in the initial hemodynamic presentation and is a major determinant of short- and long-term outcome.66 In patients without cardiopulmonary disease the anatomic and humoral obstruction must be 75% or greater to cause a mean pulmonary artery pressure (MPAP) of 40 mm Hg or higher, which is not sustainable; even the previously healthy heart may proceed to cardiovascular collapse and fail.66 Right atrial pressure (RAP) elevation occurs when the MPAP is 30 mm Hg or higher and obstruction exceeds 35% to 40%.67 Many patients who have a pulmonary embolic event have underlying cardiopulmonary disease. In these patients cardiopulmonary deterioration (CPD) may occur with a lesser degree of vascular obstruction.66 This is illustrated by the UKEP (Urokinase-Embolie Pulmonaire) Trial in which an initial presentation of shock with acute PE was seen in 56% of patients who had prior cardiopulmonary disease compared to 2% without cardiopulmonary disease. Obstruction of greater than 50% was uncommon.68 Hypoxemia is the most common gas exchange abnormality in patients. However, the PaO2 and alveolar-arterial (A-a) gradient have been reported to be normal in approximately 30% of patients with PE.69 Figure 44.2 summarizes the gas exchange abnormalities as a result of the pulmonary embolic event. Ventilation-perfusion mismatch and low mixed venous oxygen levels are the prevalent causes of hypoxemia.70,71 As pulmonary artery pressures increase, right-to-left flow across a patent foramen ovale (PFO) may occur. Endothelial damage may result from hypoxic exposure and further lead to pulmonary vasoconstriction. Hypoxemia may even promote a prothrombotic and antifibrinolytic effect.57 In acute PE an increase in alveolar dead space impairs CO2 elimination; however, the increase in minute ventilation results in hypocapnia. In patients with massive PE there can be a paradoxical elevation of the PaCO2 as a result of a marked increase in dead space ventilation.72 In PE patients because of an increase in alveolar dead space there is a reduction in alveolar CO2 content, which can be detected by capnography by a reduction in the capnographic waveform area. In one study, PE patients significantly demonstrated a reduction in the capnographic waveform area when compared to patients without PE.73 There is widening of the PETCO2-PaCO2 gradient. Bedside PETCO2 coupled with a Wells score has been shown to have a high negative predictive value.74 The signs and symptoms of acute PE are at best nonspecific. The presence of syncope, current DVT, hemoptysis, leg swelling, active cancer, surgery, leg pain, and shock each marginally increases the probability of PE. Further, the absence of dyspnea or tachycardia marginally reduces the probability of PE.75 Approximately 50% of patients with acute PE present to the emergency department. In the Emperor Registry, among 1880 documented PE patients from 22 U.S. emergency departments, the most common presenting signs and symptoms were dyspnea at rest (50%), pleuritic chest pain (39%), dyspnea with exertion (24%), and syncope, which occurred in 5% as the initial presentation of acute PE. Only 58 patients (3%) had systolic blood pressures of 90 mm Hg or lower on presentation.76 Clinical prediction rules have been used in the emergency department setting to determine the pretest probability of acute PE.77 The most widely known prediction model has been the Wells score and it can allow for a dichotomous classification.78,79 Other prediction scores have been used, such as the Geneva and the modified Geneva.77,78 All prediction scores appear to have similar accuracy but are influenced by the local prevalence of PE. The clinical prediction scores are enhanced in identifying the patient with low probability of PE coupled with low D-dimer value.79 In patients admitted to the ICU with PE the clinical manifestations may be more demonstrative, as shown in Table 44.2. In the Urokinase Pulmonary Embolism Trial (UPET), the clinical features of massive PE were evaluated. Sudden unexplained onset of dyspnea was the most common symptom and was present in 80% of the patients. A majority of the patients had a well-defined risk factor. Tachypnea and tachycardia were present in 88% and 63% of the patients, respectively. Accentuated second heart sound was noted in 67%, and S3 or S4 gallop was present in 47%. Clinical shock was noted in 12%.80 Syncope can occur. Retrosternal nonpleuritic chest pain may mimic the pain experienced with a myocardial infarction and represent demand ischemia of the right ventricle. The differential diagnosis of pulseless electrical activity (PEA) is massive PE and should be expected when there is a known major risk factor. Table 44.2 PE, pulmonary embolism; PEA, pulseless electrical activity. *Adapted from Stein PD, Willis PW III, DeMets DL: History and physical examination in acute pulmonary embolism in patients without pre-existing cardiac or pulmonary disease. Am J Cardiol 1981;47;218-223. The signs and symptoms of submassive PE will depend upon the degree of RVD. Dyspnea may be progressive over the last 4 to 6 days. Chest pain can be either pleuritic or nonpleuritic in nature. If pulmonary infarction occurs as result of distal embolization, then hemoptysis may occur. From the PIOPED II database, 92% of patients with main or lobar pulmonary emboli had dyspnea or tachycardia in contrast to segmental emboli dyspnea and tachycardia, where 65% had these findings.81,82 When a patient is admitted to the ICU for a non-PE-related clinical diagnosis, the abrupt and unexplained occurrence of some combination of chest pain, dyspnea, tachypnea, hypotension, and hypoxemia should raise the suspicion of PE. Unexplained fever with and without mild leukocytosis could represent a possible VTE event.83 Hemoptysis can occur as a result of small distal pulmonary emboli causing alveolar hemorrhage. If PE occurs while the critically ill patient or postoperative patient is on mechanical ventilation, the clinical suspicion and conformation can be difficult. An increase in minute ventilation dead space or an increase in supplemental oxygen may be clues to an embolic event. If the patient has end-tidal CO2 (PETCO2) monitoring an increase in the PaCO2-PETCO2 gradient may occur.73 Hypoxemia and an increased alveolar-arterial gradient are usually seen in PE but can be normal in about 14% and are most likely the result of small embolic events.84,85 The initial acid-base disturbance is a respiratory alkalosis, which may evolve to a metabolic (lactic) acidosis as hypotension and shock ensue. A linear relationship between PE severity and PaO2 in patients without prior cardiopulmonary disease has been described. In the UPET and PIOPED trials 12% and 19% of patients, respectively, had PaO2 80 mm Hg or greater.86 Hypocarbia is usually present with acute PE because of hyperventilation, but hypercapnia can occur with large PEs producing increased dead space.87,72 The chest radiograph is abnormal in most cases of PE; however, radiographic findings possess inadequate sensitivity to rule out PE. Common radiographic abnormalities include atelectasis, pleural effusion, parenchymal opacities, and elevation of a hemidiaphragm. Radiographic findings of pulmonary infarction often show a wedge-shaped, pleural-based triangular opacity with an apex pointing toward the hilus (Hampton hump). Focal oligemia or decreased vascularity has been known as the Westermark sign. These findings are suggestive of PE but are infrequently prospectively observed.88,89 Radiographic findings of RVD may include a prominent central pulmonary artery (knuckle sign) and cardiomegaly. In one large prospective cohort of PE patients a normal chest radiograph was found in 24% of the patients.89 A normal-appearing chest radiograph in a patient with severe dyspnea and hypoxemia, is suggestive of a pulmonary embolic event. In 1935 McGinn and White first described the association between acute PE and specific electrocardiogram (ECG) changes when they noted the S1Q3T3 pattern in seven patients with acute cor pulmonale.90 A normal ECG can occur with PE in a frequency ranging from 9% to 30%. The most common ECG abnormalities in the setting of PE are tachycardia and nonspecific ST-T wave abnormalities. The classic findings of right-sided heart strain and acute cor pulmonale are tall, peaked P waves in lead II (P pulmonale); right-axis deviation; right bundle branch block; and an S1Q3T3 pattern. Only 20% of patients with proven PE have any of these classic ECG abnormalities.91–93 T-wave inversion in the precordial leads representing subepicardial ischemia may be seen. The presence of this ECG finding is associated with increased degree of vascular obstruction and elevation of the pulmonary artery (PA) pressure.94 The new onset of atrial fibrillation has been thought to be associated with a pulmonary embolic event. In a recent study the occurrence of atrial fibrillation did not in general increase the probability of PE.95 However, if chest pain were the only symptom, the presence of atrial fibrillation increases the probability of PE (odds ratio [OR] 2.42, 95% confidence interval [CI] 0.97-6.07). The serum D-dimer is a degradation product produced by plasmin-mediated proteases of cross-linked fibrin. D-dimer is measured by a variety of assays. These assays are categorized as quantitative, semiquantitative, qualitative rapid enzyme-linked immunosorbent assays (ELISAs); quantitative and semiquantitative latex; and whole-blood assays. The quanitative rapid ELISAs are the best assays to use in terms of sensitivity and likelihood ratio.96 When clinical prediction scores indicate that the patient has a low or moderate pretest probability of PE, D-dimer testing is useful to further define the likelihood of PE. Negative results on a high-sensitivity D-dimer test in a patient with a low pretest probability of PE (Wells rule) are associated with a low likelihood of VTE and reliably exclude PE.97 In a large prospective, randomized trial in patients with a low probability of PE who had negative D-dimer results, not performing additional diagnostic testing was not associated with an increased frequency of symptomatic VTE during the subsequent 6 months.98 D-dimer testing loses its ability to predict the likelihood of PE in older patients and in the presence of cancer, pregnancy, and various inflammatory or infectious disorders. If the pretest probability for PE is high, D-dimer testing should not be performed and objective testing should be done.96 Combining D-dimer results with measurement of the exhaled end-tidal ratio of carbon dioxide to oxygen (PETCO2/PO2) can be useful for diagnosis of PE. Kline and colleagues demonstrated in moderate-risk patients a positive D-dimer (>499 ng/mL) and a PETCO2/PO2 less than 0.28 significantly increased the probability of finding segmental or larger PE on computed tomography (CT) multidetector-row pulmonary angiography.99 In contrast, a PETCO2/PO2 greater than 0.45 predicted the absence of segmental or larger PE. Currently, DVT is largely diagnosed by duplex CUS. When using CUS, the ultrasonographic transducer pressed against veins shows they are easily compressed, but the muscular arteries are extremely resistant to compression. Where DVT is present, the veins do not collapse completely when pressure is applied using the ultrasonographic probe.100–102 Before an ultrasonographic scan can be considered negative, the entire deep venous system must be examined using centimeter-by-centimeter compression testing of every vessel. The risk of a DVT event following a single whole leg CUS in 3 months is 0.57% (95% CI 0.25-0.89).103 CUS is associated with a sensitivity of 97% for proximal DVT, 72% for distal DVT, and a specificity of 94%.104 In a meta-analysis of the role of CUS in DVT, CUS alone had a sensitivity of 93.8% (92.0-95.3%) for proximal DVT, 56.8% (49.0-66.4%) for distal DVT, and specificity of 97.8% (97.0-98.4%).105 The A high-probability scan, as shown in Figure 44.3, is sufficient diagnostic evidence of PE to begin anticoagulation therapy, and a normal Preexisting cardiopulmonary disease diminishes the clinical utility of The PIOPED II study firmly established the role of CT pulmonary angiography (CTPA) and largely replaced ventilation-perfusion ( In the PIOPED II study, CT angiography of the chest, as many as 25% of patients had a contraindication for the test. Preexisting renal failure and pregnancy were the primary conditions that prevented this mode from being used as the primary test for the evaluation of PE.110 Thus, gadolinium-enhanced magnetic resonance pulmonary angiography (MRA) could potentially be used to diagnose PE because it is devoid of radiation. The accuracy of this technique combined with magnetic resonance venography (MRV) has been studied in the prospective, multicenter PIOPED III accuracy study. The proportion of technically inadequate images ranged from 11% to 52% across the seven participating centers. Technically adequate MRA had a sensitivity of 78% and a specificity of 99%, whereas technically adequate MRA and MRV had a sensitivity of 92% and a specificity of 96%. However, 194 (52%) of 370 patients had technically inadequate results, which substantially restricts its clinical use.114 Death from an embolic event almost always results from hemodynamic deterioration due to an abrupt rise in pulmonary vascular resistance overwhelming the contractile power of the right ventricle. Progressive hemodynamic and respiratory deterioration characterizes the clinical presentation of major or massive PE and is seen in approximately 5% of PE patients.2 Patients can be categorized as having submassive PE. Risk stratification becomes important to identify this group of patients who present with relative hemodynamic stability but exhibit varying degrees of RVD. Risk stratification schemes in normotensive patients center on RV function assessment after an embolic event and identify patients who may be at risk for progressive hemodynamic deterioration. Approximately 50% of patients who are normotensive have varying degrees of RVD and 10% will die. The European Society of Cardiology recommends a classification of PE into high-risk and non–high-risk groups. The high-risk groups represent the massive or major PE presentations. The non–high-risk group is further subdivided into intermediate risk in which the mortality rate ranges between 3% and 15% and a low-risk group in which the mortality rate is less than 3%. Initial therapeutic interventions are greatly influenced by risk stratification that includes clinical scoring systems, laboratory data, and imaging.115–117 The presence of RVD is a predictor of the increased risk of embolic-related adverse events and mortality rates when compared with patients without RVD.2 Patients who present with circulatory shock usually have severe RVD and have mortality rates up to 65%.1,2,115 In PE patients with normal hemodynamics and evidence of RVD the mortality rates can range from 8% to 14%. Several registry studies have observed that the frequency of RVD in normotensive patients is approximately 40%.116,117 In PE patients with initial normal hemodynamics and no RVD the mortality rate is low (0-3%). The initial hemodynamic presentation of PE is the most powerful predictor of outcome, as illustrated in Figure 44.5. Arterial hypotension at the time of PE presentation is an early predictor of death. In the 2392 patients from the ICOPER study, the 90-day mortality rates were 52.4% in patients with systolic arterial blood pressures below 90 mm Hg, and 14.7% in those patients who had preserved arterial blood pressures. In addition, comorbid conditions increase the risk of adverse clinical events, even in the presence of anatomically small PE. In the ICOPER findings, age over 70, congestive heart failure, cancer, and chronic lung disease were identified as independent predictors of 3-month mortality risk, with approximately twofold increase in the risk of death.2 In one study, altered mental status at presentation was associated with a sevenfold increase in risk of adverse outcome. Altered mental status most likely reflected decreased cardiac perfusion or hypoxia. In the same study the initial presentation of shock was associated with a threefold increase in risk of 30-day adverse outcome (death, secondary shock, or recurrent PE).117 Patients can be clinically risk stratified by the use of a clinical scoring system known as the Pulmonary Embolism Severity Index (PESI), which also has a simplified version.118,119 This prognostic model was developed by Aujesky and colleagues (Table 44.3) using a large database of hospitalized PE patients from which they derived and validated a prediction rule that identified acute PE patients with low to high risk for fatal PE and adverse events. Using 11 variables of predictors, five distribution categories emerged looking at inpatient fatality, 30-day all-cause mortality rate, and the adverse outcomes of nonfatal cardiogenic shock or cardiopulmonary arrest. Class I to class III all-cause mortality rates ranged between 0% and 3.1%, although class IV to class V all-cause mortality rates were between 10% and 24%. This score has an excellent negative predictive value and readily identifies low-risk patients but suffers in the attempt to predict patients at high risk for adverse events.119 A more simplied PESI score has been developed and has a high negative prediction value.120 Table 44.3 Pulmonary Embolism Severity Index (PESI) Adapted from Aujesky D, Obrosky DS, Stone RA, et al: Derivation and validation of a prognostic model for pulmonary embolism. Am J Respir Crit Care Med 2005;172:1041-1046. In patients with symptomatic acute PE, the presence of DVT may have prognostic significance. In a prospective study by Jiménez and coworkers, among 707 patients diagnosed with acute PE, 51.2% had concomitant DVT and 10.9% had PE-specific fatality at a 3-month follow-up. Patients with PE with the presence of DVT at the time of diagnosis had an adjusted hazard ratio (HR) of 2.05% (95% CI 1.24 to 3.38) and PE-specific fatality-adjusted HR of 4.25 (95% CI 1.61 to 11.25). These findings were validated by a large international registry trial (RIETE) that again demonstrated a significant prediction of all-cause mortality rate and PE-specific mortality rate when the concomitant DVT was present during the acute embolic event. These results suggest that patients with symptomatic acute PE have an increase in all-cause mortality rate, PE-related death, and recurrent VTE over a 3-month period if at the time of PE a diagnosis of proximal DVT is also present.121 Several studies have demonstrated that ECG signs of RV strain correlate with the presence of RVD.122,123 A study by Punukollu and associates showed that T-wave inversion in leads V1 to V3 had a specificity of 88% and diagnostic accuracy of 81% for RVD in acute PE.124 Daniel and colleagues developed an ECG scoring system based on typical features of acute PE such as presence of right bundle branch block (RBBB), T-wave inversions, and additional features of right-sided heart strain that predicted severe pulmonary hypertension (sPAP >50 mm Hg) with a specificity of 97.7% if the patient’s “ECG score” is 10 or greater.125 The ECG may also be a tool for risk stratification of normotensive acute PE patients. In a study by Vanni and coworkers among 306 patients with documented PE 130 (34%), patients demonstrated ECG findings of right-sided heart strain. In this study ECG findings of right-sided heart strain were defined as incomplete or complete RBBB, S1Q3T3, or inverted T waves in the precordial leads V1, V2, and V3. For this study only one of these ECG patterns had to be present. The investigators demonstrated that the presence of RV strain on the initial ECG was associated with clinical deterioration and death (HR 2.58) in normotensive PE patients.122 The echocardiogram has become the main imaging modality to assess RV structure and function. In addition, pulmonary artery pressures can be estimated. Most hospitals in the United States have portable echocardiographic technology allowing for serial assessment of the impact of treatment on RVD. In hemodynamically stable patients with PE, 20% to 40% will have some echocardiographic finding of RVD.116,117 Box 44.1 illustrates the echocardiographic finding of acute PE. Toosi and colleagues performed a quantitative risk assessment of various echocardiographic measurements of RVD in acute PE. For in-patient mortality rate the positive predictive values range between 11% and 20% and the negative predictive values are 97% to 100%.140 The major drawback with the role of the echocardiographic definition of RVD of acute PE is that it lacks standardization. There is no clear agreement on which of the echocardiographic The heart will release natriuretic peptides as result of ventricular volume overload. Ventricular stretch results in cellular production of proBNP. This peptide is subsequently cleaved to the biologically active BNP and the inactive N-terminal fragment (NT-proBNP).126 Both BNP and NT-proBNP can be measured clinically and have been validated as diagnostic and prognostic markers of ventricular stretch in multiple conditions, including congestive heart failure, pulmonary hypertension, and acute PE.127 Elevated concentrations of BNP distinguish patients with PE at higher risk of complicated in-hospital course and death from those with low BNP levels. Both BNP and NT-proBNP have been extensively investigated as prognostic markers in acute PE. In an early study, ten Wolde and colleagues127 demonstrated in a prospective cohort of normotensive acute PE patients that those who died within 90 days had a significantly increased BNP level at the time of diagnosis compared with those patients who did not die in follow-up (245 pg/mL vs. 30 pg/mL, respectively). In addition, using a cut-off point of 75 pg/mL, these investigators reported that BNP had a positive predictive value of PE-related fatality of 17%, whereas the negative predictive value for death was 99%.127 As will be seen for most of the prognostic markers for acute PE, the investigators noted that BNP was able to identify those at low risk of adverse outcomes but did not have good specificity (many false-positives) in identifying those who were likely to die from their PE. Normal levels of BNP have a high negative predictive value for an elevated level of BNP or NT-proBNP as a risk factor for short-term fatality and overall short-term complicated clinical outcome, and an indicator of RVD in patients with acute PE. This is illustrated by the study by Kucher and colleagues,128 who investigated the prognostic capabilities of NT-proBNP in a prospective cohort of acute PE patients. In those suffering an adverse event (defined as a combined end point of in-hospital death or need for escalation of care, including cardiopulmonary resuscitation, mechanical ventilation, vasopressor, or thrombolysis), NT-proBNP was significantly higher compared with those who did not experience an adverse event (median of 4250 pg/mL vs. 121 pg/mL, respectively). An NT-proBNP cut-off point of 500 pg/mL yielded a negative predictive value for adverse outcome of 97% with a corresponding positive predictive value of adverse outcome of 45%.128 In a meta-analysis of 13 studies by Klok and associates, they indeed demonstrate that increased BNP or proBNP was a risk factor for short-term fatality and adverse clinical events and a marker for RVD. However, the investigators caution against using this biomarker alone to justify more aggressive medical therapy, such as thrombolytic therapy.129 Serum troponins are widely available for the evaluation of diverse forms of myocardial injury. The assays are rapid and are well suited as a risk stratification tool. A number of studies and meta-analyses have evaluated the prognostic value of cardiac troponin I (TnI) or T (TnT) (determined by conventional assays) in acute PE.130
Acute Pulmonary Embolism
Prevalence of Venous Thromboembolism in Intensive Care Unit Patients
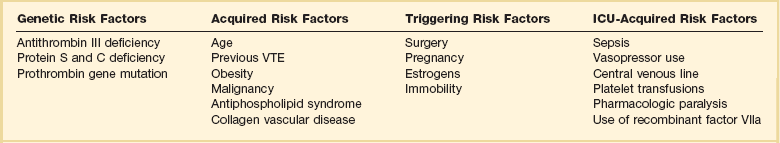
Risk Factors
 Virchow has described several risk factors unique to the ICU setting that predispose patients to VTE events. In a prospective observational study of 93 medical-surgical ICU patients, the investigators identified several ICU-related factors that are associated with increased risk of VTE, including mechanical ventilation, immobility, femoral venous catheters, sedatives and paralytic drugs, and failure to prescribe VTE prophylaxis. Prolonged mobility in the ICU setting is the result of mechanical ventilation, sedative administration, and paralytic agents. These four factors (mechanical ventilation, immobility, sedation, and paralysis) may converge to greatly increase the risk of VTE.26 Sedation and paralysis also predispose to other ICU complications including increased duration of mechanical ventilation, and critical illness polyneuropathy.27,28
Virchow has described several risk factors unique to the ICU setting that predispose patients to VTE events. In a prospective observational study of 93 medical-surgical ICU patients, the investigators identified several ICU-related factors that are associated with increased risk of VTE, including mechanical ventilation, immobility, femoral venous catheters, sedatives and paralytic drugs, and failure to prescribe VTE prophylaxis. Prolonged mobility in the ICU setting is the result of mechanical ventilation, sedative administration, and paralytic agents. These four factors (mechanical ventilation, immobility, sedation, and paralysis) may converge to greatly increase the risk of VTE.26 Sedation and paralysis also predispose to other ICU complications including increased duration of mechanical ventilation, and critical illness polyneuropathy.27,28
Deep Venous Thrombosis
Pathophysiology
Hemodynamic Consequences
Respiratory Consequences
Clinical Presentation
Clinical Presentation on Admission to the Intensive Care Unit
Recognition of Pulmonary Embolism During Intensive Care Unit Admission
Diagnostic Testing for Pulmonary Embolism
Arterial Blood Gas Measurement
Chest Radiography
Electrocardiography
D-Dimer Assay
Duplex Compression Ultrasonography
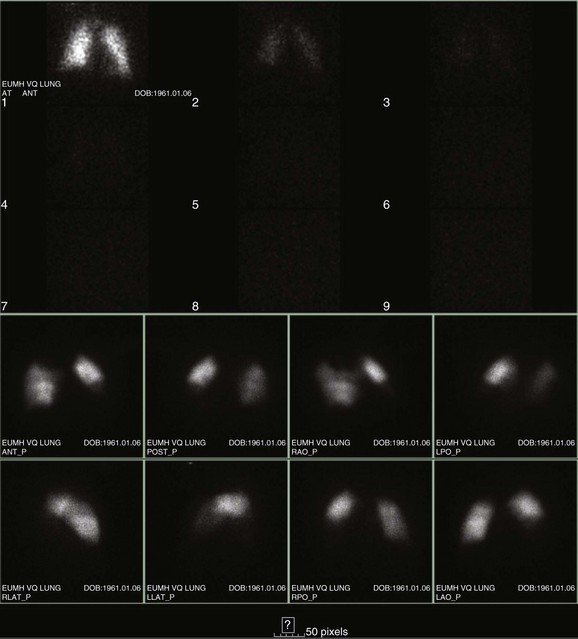
Ventilation-Perfusion Nuclear Scans
 scan has a limited role in the diagnosis of acute PE in the ICU, although its diagnostic accuracy is no different from that in non–critically ill populations.106 Because of the advancing technology of CT scanning, the lung scan is used in the critical care units in special circumstances such as renal insufficiency or in the patient who is not intubated and capable of transport from the ICU to the nuclear medicine suite for a full view scan. If the institution has a portable lung scanner this can be used in the ICU but the number of images is reduced when compared to the standard
scan has a limited role in the diagnosis of acute PE in the ICU, although its diagnostic accuracy is no different from that in non–critically ill populations.106 Because of the advancing technology of CT scanning, the lung scan is used in the critical care units in special circumstances such as renal insufficiency or in the patient who is not intubated and capable of transport from the ICU to the nuclear medicine suite for a full view scan. If the institution has a portable lung scanner this can be used in the ICU but the number of images is reduced when compared to the standard  scan.
scan.
 scan is considered sufficient evidence to exclude PE. However, the frequency of low- or intermediate-probability scans (indeterminate scans) can be as high as 50% to 70%, with a 10% to 50% probability of PE. Therefore, it is impossible to initiate anticoagulation therapy based on this indeterminate lung scan probability. In the first PIOPED study, only 40% of patients with PE had a high-probability
scan is considered sufficient evidence to exclude PE. However, the frequency of low- or intermediate-probability scans (indeterminate scans) can be as high as 50% to 70%, with a 10% to 50% probability of PE. Therefore, it is impossible to initiate anticoagulation therapy based on this indeterminate lung scan probability. In the first PIOPED study, only 40% of patients with PE had a high-probability 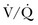 scan result, whereas another 40% of patients with PE had an indeterminate result and 14% had a low-probability result. A high-probability
scan result, whereas another 40% of patients with PE had an indeterminate result and 14% had a low-probability result. A high-probability 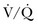 scan had a positive predictive value of 88% in patients without prior history of PE. The combination of a high clinical probability with the high-probability lung scan resulted in a 96% positive predictive value. The negative predictive value of a low-probability scan was 88% and improved to 96% when combined with a low clinical probability. Thus, a low-probability scan with a low clinical probability score rules out PE as definitively as a normal scan. Finally, 39% of the patients had intermediate scans and 32% of these patients had angiographically proven PE.14,107
scan had a positive predictive value of 88% in patients without prior history of PE. The combination of a high clinical probability with the high-probability lung scan resulted in a 96% positive predictive value. The negative predictive value of a low-probability scan was 88% and improved to 96% when combined with a low clinical probability. Thus, a low-probability scan with a low clinical probability score rules out PE as definitively as a normal scan. Finally, 39% of the patients had intermediate scans and 32% of these patients had angiographically proven PE.14,107
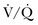 scans by increasing the prevalence of nondiagnostic scans. Nondiagnostic scans are more likely to occur in patients with preexisting cardiopulmonary disease. Lung scans are not useful to either establish or rule out the diagnosis of PE in more than 80% of cases. A similar disadvantage of
scans by increasing the prevalence of nondiagnostic scans. Nondiagnostic scans are more likely to occur in patients with preexisting cardiopulmonary disease. Lung scans are not useful to either establish or rule out the diagnosis of PE in more than 80% of cases. A similar disadvantage of 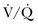 lung scans is noted in critically ill patients with prior cardiopulmonary disease. Nevertheless,
lung scans is noted in critically ill patients with prior cardiopulmonary disease. Nevertheless, 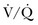 lung scans can be performed in the critically ill, even if patients are receiving mechanical ventilation, and chest radiographic abnormalities can be used as a surrogate of ventilation defects. Despite the limited ventilation component under positive-pressure respiration, the perfusion component is not significantly affected by mechanical ventilation.
lung scans can be performed in the critically ill, even if patients are receiving mechanical ventilation, and chest radiographic abnormalities can be used as a surrogate of ventilation defects. Despite the limited ventilation component under positive-pressure respiration, the perfusion component is not significantly affected by mechanical ventilation. 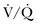 scans remain a useful tool in patients with contraindications to pulmonary angiography or CT angiography.108,109
scans remain a useful tool in patients with contraindications to pulmonary angiography or CT angiography.108,109
Multidetector Computed Tomography Scan of the Chest
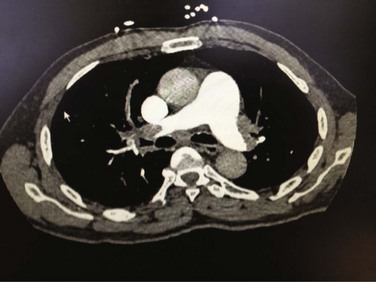
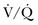 ) lung scintigraphy as the main imaging modality in suspected PE.110 The technology is continually evolving and its use is widespread. As illustrated in Figure 44.4, CTPA provides direct visualization of emboli throughout the pulmonary arterial vasculature. The advantage of CTPA over pulmonary angiography and lung scan is the discovery of an alternative diagnosis when PE is not found on CTPA. In one study, an alternative diagnosis was established in 57% of patients who did not have a PE.111 Currently, CTPA is the “gold standard” for PE diagnosis. A meta-analysis of 23 studies with 4657 patients with a negative CTPA who did not receive anticoagulation showed a 3-month rate of subsequent VTE of 1.4% (95% CI 1.1-1.8) and a 3-month rate of fatal PE of 0.51% (0.33-0.76), which compares favorably with the results noted after a normal “gold standard” invasive contrast pulmonary angiogram.112 The increased use of CTPA raises concerns about the increased incidence of cancer attributable to radiation.113 For this reason, combined use of CTPA and CT leg venography should not be performed because the latter adds little to diagnostic utility. In the PIOPED II trial, no patient with PE or DVT would have been undiagnosed if imaging of the pelvic and proximal leg veins had been omitted.107,110
) lung scintigraphy as the main imaging modality in suspected PE.110 The technology is continually evolving and its use is widespread. As illustrated in Figure 44.4, CTPA provides direct visualization of emboli throughout the pulmonary arterial vasculature. The advantage of CTPA over pulmonary angiography and lung scan is the discovery of an alternative diagnosis when PE is not found on CTPA. In one study, an alternative diagnosis was established in 57% of patients who did not have a PE.111 Currently, CTPA is the “gold standard” for PE diagnosis. A meta-analysis of 23 studies with 4657 patients with a negative CTPA who did not receive anticoagulation showed a 3-month rate of subsequent VTE of 1.4% (95% CI 1.1-1.8) and a 3-month rate of fatal PE of 0.51% (0.33-0.76), which compares favorably with the results noted after a normal “gold standard” invasive contrast pulmonary angiogram.112 The increased use of CTPA raises concerns about the increased incidence of cancer attributable to radiation.113 For this reason, combined use of CTPA and CT leg venography should not be performed because the latter adds little to diagnostic utility. In the PIOPED II trial, no patient with PE or DVT would have been undiagnosed if imaging of the pelvic and proximal leg veins had been omitted.107,110
Magnetic Resonance Imaging
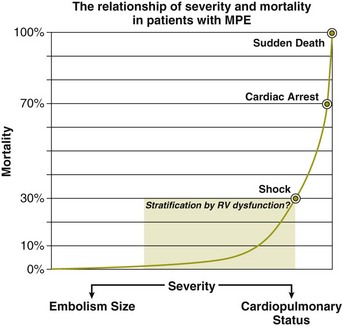
Risk Stratification
Clinical and Hemodynamic Parameters
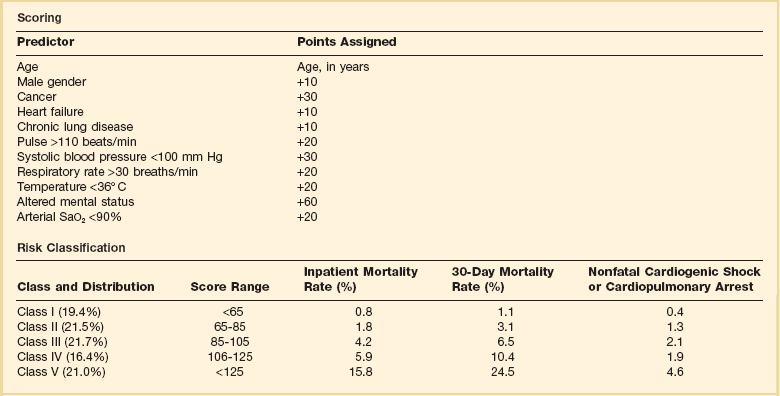
Electrocardiogram
Cardiac Biomarkers
 Biomarkers are relied on in the risk stratification of patients with PE. Tests for biomarkers are widely available and there is usually a quick turnaround from the clinical laboratory. Caution must be taken on the interpretation of these biomarkers. It is important for the critical care clinician to understand the clinical issues that impact the interpretation of the biomarkers and understand the negative and positive predictive values.115 Elevations of brain natriuretic peptide (BNP) and troponin occur in many patients with PE and correlate with magnitude of RVD.
Biomarkers are relied on in the risk stratification of patients with PE. Tests for biomarkers are widely available and there is usually a quick turnaround from the clinical laboratory. Caution must be taken on the interpretation of these biomarkers. It is important for the critical care clinician to understand the clinical issues that impact the interpretation of the biomarkers and understand the negative and positive predictive values.115 Elevations of brain natriuretic peptide (BNP) and troponin occur in many patients with PE and correlate with magnitude of RVD.
Echocardiography
Brain Natriurectic Peptide
Cardiac Troponin
Related posts:

Full access? Get Clinical Tree


Acute Pulmonary Embolism