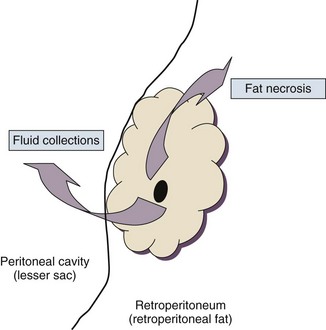
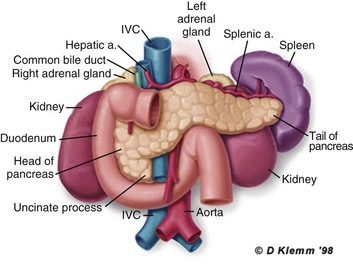
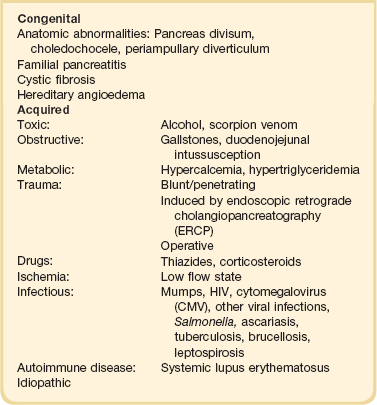
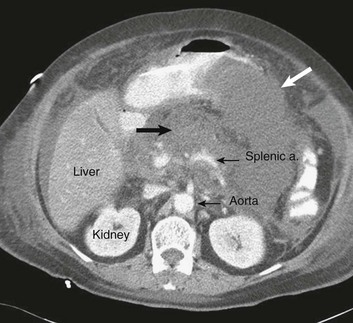
77 CLINICAL PRESENTATION AND DIAGNOSIS EARLY MANAGEMENT OF THE CRITICALLY ILL PATIENT WITH ACUTE PANCREATITIS MANAGEMENT OF THE LATE COMPLICATIONS OF SEVERE ACUTE PANCREATITIS LONG-TERM OUTCOME AND QUALITY OF LIFE Acute pancreatitis is a complex disease with a highly variable clinical course. It is responsible for more than 200,000 hospital admissions each year in the United States,1 and it has an annual incidence of 10 to 80 cases in 100,000 in the developed world.2–5 Incidence rates have been increasing.6 For reasons that are unknown, there is seasonal variation in rates of the disease, the incidence being maximal in the spring and fall.7 The crude mortality rate for patients who are hospitalized with acute pancreatitis is less than 2%,8 and the majority of patients with acute pancreatitis experience a benign and self-limited disease, resulting in a hospital stay of only several days and no significant lasting sequelae. In a small percentage of patients, however, acute pancreatitis is sufficiently severe to lead to admission to an intensive care or high dependency unit, and a complicated clinical course with a mortality risk that may exceed 20%.1,9 These latter patients can present the intensivist with formidable challenges during the course of their illness, but if they recover, as most do, they can return to their premorbid state of health with no significant diminution in quality of life. The prognosis for patients with severe acute pancreatitis has improved considerably since Ranson proposed his widely used severity criteria. In the mid-1970s, the mortality for patients with severe pancreatitis approached 100%10; today the risk is less than one quarter of that figure.11,12 This improved outcome can be ascribed in part to general improvements in the care of critically ill patients and, more important, to fundamental changes in the approach to the medical and surgical care of the patient with acute pancreatitis that, in turn, reflect an evolving understanding of the pathophysiology of the disease. Pancreatitis is an acute inflammatory disorder that arises as a consequence of the activation of pancreatic digestive enzymes within the parenchyma of the gland and the surrounding tissues of the peritoneal cavity and retroperitoneum. Its evolution and complications are variable and give rise to terminology that is both confusing and imprecise. The most widely used classification system is that known as the Atlanta classification from 199213 (Table 77.1). Its reproducibility, however, is poor,14 and it is easier to conceptualize the disease as a spectrum of overlapping abnormalities resulting from the leakage of activated pancreatic enzymes, the host response to local tissue injury, and the superimposed complication of infection of what is initially a sterile process (Fig. 77.1). Table 77.1 The Atlanta Classification System for Acute Pancreatitis Adapted from Bradley EL: A clinically based classification system for acute pancreatitis. Arch Surg 1993;128:586-590. In the mildest form of the disease, leakage of activated enzymes is minimal, and the most prominent manifestation is pancreatic edema secondary to a local inflammatory process. In more severe cases, pancreatic ductal disruption results in leakage of pancreatic enzymes into adjacent tissues. If the leakage occurs anteriorly into the peritoneal cavity, fluid collections will be evident. The local peritoneal inflammatory response triggers coagulation and fibrin deposition, walling the collection off and creating a pseudocyst, so called because it is a fluid collection that lacks an epithelial lining. Drainage of the pancreatic ascites through the diaphragm can create a pleural effusion; typically this is seen on the left. Marked elevation of the amylase level in the pleural fluid establishes the collection as pancreatic juice, rather than as a reaction to an inflammatory process on the abdominal side of the diaphragm. Alternatively, if leakage is into the fatty tissues of the retroperitoneum, necrosis predominates, although small loculated fluid collections are often present. Often in patients with severe acute pancreatitis, computerized tomography shows evidence of both intraperitoneal and retroperitoneal involvement. The older term, hemorrhagic pancreatitis, describes the sequelae of extension of retroperitoneal necrosis into blood vessels of the retroperitoneum. Extraperitoneal tracking of the resulting hematoma gives rise to Grey-Turner’s sign when the ecchymosis is evident in the flanks and Cullen’s sign when it tracks anteriorly through the falciform ligament to present at the umbilicus.15 Acute pancreatitis arises in the pancreas through the leakage of activated pancreatic enzymes into pancreatic and peripancreatic tissues. The characteristic clinical syndrome, however, reflects the activation of a massive systemic inflammatory response to that local tissue injury, mediated through the activation of the inflammasome—a multiprotein intracellular complex that leads to the activation of the key inflammatory cytokine, interleukin-1.16 Beyond its role as an endocrine organ, the pancreas plays a fundamental role in the digestion of foodstuffs through the production of enzymes that degrade the major constituents of ingested food: protein (proteases), fat (lipases), starch (amylase), and nucleic acids (nucleases). Pancreatic enzymes are synthesized by the acinar cells lining the pancreatic ductal system and released from the cell as inactive zymogens.17 They pass via the pancreatic duct into the lumen of the duodenum, where they are activated by mucosal enzymes such as enterokinase, and so become capable of degrading their target substrates. Under normal circumstances, activation is vigorously inhibited within the pancreas itself through sequestration of newly synthesized enzymes within zymogen granules, and further through the local action of a specific inhibitor of trypsin activation called SPINK.18 The importance of normal mechanisms that inhibit trypsin activation is underlined by the observation that genetic mutations in the SPINK gene associated with reduced activity have been implicated in the pathogenesis of hereditary pancreatitis.19 Acute pancreatitis arises through the activation of pancreatic enzymes within the pancreas itself, possibly through the activity of lysosomal hydrolases in the acinar cell and initiating autodigestion of the pancreas and surrounding tissues.20,21 Activation of trypsinogen to trypsin appears to be a critical event during the early pathogenesis of the acute process,22,23 and an increase in intracellular calcium concentrations may contribute to activation of trypsinogen.24 Trypsinogen can also be activated by lysosomal cathepsin B within the acinar cell.25 Pancreatic acinar cells can die by either necrosis or apoptosis. Necrotic death is characterized by cell lysis, with the leakage of intracellular constituents into the surrounding microenvironment and the activation of an acute inflammatory response. Neutrophil recruitment in response to the local tissue injury has been implicated in the amplification of cellular damage in acute pancreatitis.26–28 Apoptotic death, in contrast, is noninflammatory. Cells are degraded into membrane-bound vesicles that are taken up by fixed tissue macrophages. Phagocytosis of an apoptotic cell not only prevents the local activation of inflammation but triggers transcriptional programs in the phagocytosing cell that are anti-inflammatory and reparative in nature, with up-regulation of counter-inflammatory cytokines such as interleukin-10 (IL-10).29 The local cellular injury in acute pancreatitis can also be conceptualized as an imbalance between necrotic and apoptotic cell death. Activation of caspases—the intracellular enzymes that mediate apoptosis—attenuate the severity of pancreatitis,30 whereas severe pancreatitis is associated with reduced levels of IL-10.31 Activated pancreatic enzymes injure not only cells of the pancreas but also those of surrounding tissues. Fat cells appear to be particularly vulnerable, and the disease tends to be more severe in the obese because of the greater amount of peripancreatic fat necrosis.32 However, the degradative effects of pancreatic enzymes, and the secondary tissue injury resulting from the host response, can injure other structures in the vicinity of the pancreas, in particular, the transverse colon, and major vessels such as the splenic artery and vein.33 Pancreatitis rapidly evolves from an inflammatory disease of the pancreas to a chemical burn of the retroperitoneum. The pathogenesis of the clinical syndrome is further complicated by the presence of the gastrointestinal tract immediately adjacent to the pancreas (Fig. 77.2). The head of the pancreas lies within the curve of the duodenum, immediately behind the stomach and above the transverse colon. Damage to terminal feeding vessels in the colonic fat caused by activated pancreatic enzymes can result in focal perforation of the colon, with leakage of colonic bacteria into the peritoneal cavity.34 Although the stomach and duodenum are only lightly colonized in health, the local ileus induced by the acute pancreatic inflammation promotes proximal gut overgrowth with enteric bacteria.35 In addition, changes in gut mucosal barrier function arising from local inflammation and the absence of enteral nutrients promote the translocation of luminal bacteria and of bacterial products such as endotoxin into the injured or necrotic peripancreatic tissues.36 Ultimately in the more severe cases, the clinical syndrome evolves as a result of a systemically activated inflammatory response, with the same changes in microvascular blood flow, endothelial permeability, and inflammatory mediator release that characterize bacterial sepsis.37 Acute pancreatitis has many causes, although 80% of cases in the developed world result from either alcohol or gallstones (Table 77.2). The mechanism of alcohol-induced pancreatitis is unclear. The acinar cell of the pancreas is capable of metabolizing alcohol and also appears to be the primary target of alcohol-mediated pancreatic injury.38 In vitro studies show that alcohol reduces the sensitivity of isolated acini to zymogen activation by cholecystokinin.39 Gallstone pancreatitis appears to be a consequence of a transient acute increase in pancreatic ductal pressures associated with passage of a small gallstone through the sphincter of Oddi in patients with a common channel for the bile and pancreatic ducts.40 True obstruction of the duct is uncommon, and approximately 90% of patients will be found to have gallstones in the stool, implicating the passage of the stone, rather than an obstructing mechanism, in the etiology of the resulting pancreatic inflammation. The list of drugs implicated in the etiology of acute pancreatitis is long41; some of the more prominent associations are shown in Table 77.2. Genetic factors have been associated with the development of acute pancreatitis. A polymorphism in the secretory trypsin inhibitor (SPINK1) gene is associated with an increased risk for acute pancreatitis,42 as are mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene,43 and PRSS1.44 Polymorphisms in the genes for tumor necrosis factor α (TNFα) and heat shock protein 70 have also been linked to an increased risk for acute pancreatitis,45 although the association is less clear. The causes of shock in the patient with acute pancreatitis are multifactorial. Initially, the acute inflammatory process in the retroperitoneum elicits local inflammation, with an outpouring of fluid into the relatively confined space of the retroperitoneum or into the peritoneal cavity itself. Intra-abdominal inflammation evokes secondary ileus within the gastrointestinal tract, and fluid is sequestered here, increasing the relative intravascular volume deficit. Nausea, vomiting, and a reluctance to take fluids by mouth further exacerbate this fluid deficit. As the process evolves, a systemic inflammatory response to the local abdominal process results in diffuse vasodilatation and capillary leak syndrome. In aggregate, the effective loss of intravascular volume can be enormous, and so the initial resuscitation may require large volumes of intravenous fluid. Acute hypocalcemia may also contribute to the clinical picture of cardiovascular compromise. Indeed the classical prognostic criteria articulated by Ranson (Table 77.3) emphasize the importance of acute inflammation (white cell count), and the secondary hemodynamic (fluid sequestration, base excess, blood urea nitrogen [BUN], and hematocrit) and metabolic (glucose, calcium) sequelae in determining the ultimate prognosis. Table 77.3 Ranson’s Criteria in Acute Pancreatitis LDH, Lactate dehydrogenase; AST, aspartate aminotransferase; BUN, blood urea nitrogen. From Ranson JH, Rifkind KM, Roses DF, et al: Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet 1974;139:69-81. The diagnosis of pancreatitis is usually straightforward, based on the combination of clinical manifestations and characteristic biochemical findings of elevations in the circulating levels of amylase and lipase, the latter being somewhat more accurate diagnostically.46 The diagnosis can be confirmed, and the severity of the disease evaluated by computerized tomography (Fig. 77.3), using the grading system developed by Balthazar47 (Table 77.4). Table 77.4 Balthazar Grading of CT Findings in Acute Pancreatitis From Balthazar EJ: CT diagnosis and staging of acute pancreatitis. Radiol Clin North Am 1989;27:19-37. An initial assessment of the severity of the disease is useful primarily for deciding the optimal venue for the early management of the patient, as at least some of the delayed morbidity of acute pancreatitis can be reduced through aggressive initial resuscitation and support. Various approaches have been used to quantify disease severity. Ranson identified 11 variables—5 at initial presentation and 6 over the ensuing 48 hours—that correlated in a graded fashion with the ultimate risk of mortality (see Table 77.3). The Glasgow-Imrie criteria are a modification of Ranson’s scale and represent an alternate model of severity scoring. In head-to-head studies, Acute Physiology, Age, and Chronic Health Evaluation (APACHE) II—a generic severity of illness scale—performs at least as well as the Ranson or Glasgow-Imrie criteria in predicting hospital survival.48 Moreover, even simpler scales appear to provide comparable prognostic information.23,49 Various biochemical measures, including C-reactive protein, procalcitonin, interleukin-6, and trypsinogen activation peptide (TAP), are purported to differentiate mild and severe pancreatitis early during the clinical evolution of the disease50–52; their clinical utility is unclear. Persistence of clinical manifestations of systemic inflammation beyond 48 hours is also associated with subsequent organ dysfunction and a higher mortality.53 Precision in prognostication is far less important than early recognition of the patient for whom close monitoring and aggressive resuscitation can alter the clinical course, and a low threshold for management in a more controlled and monitored setting is an important factor in reducing the complications of pancreatitis.54 Intravascular fluid deficits early in the course of pancreatitis can be substantial. Early aggressive fluid resuscitation is the cornerstone of initial successful management.55 Volume status should be monitored with a urinary catheter and central venous catheter, and although studies in the specific setting of pancreatitis have not been performed, there is every reason to believe that the principles of goal-directed resuscitation should be followed.56 There are no data to support a preference for colloids or crystalloids during resuscitation; the key, however, is to administer sufficient volumes rapidly enough to restore an adequate circulating volume.57 Typically this comprises many liters of fluid, and not infrequently, the sickest patients will receive more than 10 to 15 L of fluid resuscitation over the first 24 hours. Such aggressive resuscitation is best carried out within the well-monitored environment of the intensive care unit (ICU). Although aggressive fluid resuscitation can minimize later complications such as pancreatic necrosis and acute renal failure, in the patient with increased vascular permeability it carries a high risk of complications related to interstitial edema. Frequently patients will require endotracheal intubation and mechanical ventilation. Development of the abdominal compartment syndrome is a relatively common and underdiagnosed complication of resuscitated acute pancreatitis.58 Intra-abdominal pressure can be measured by transducing a urinary catheter in the bladder. Normal pressures approximate the central venous pressure, whereas pressures greater than 20 cm H2O indicate intra-abdominal hypertension, and pressures greater than 30 cm H2O indicate a compartment syndrome and carry an increased risk of ischemic injury because of impairment of visceral venous drainage. In severe cases, management requires abdominal decompression by laparotomy. The need for laparotomy in the patient with acute pancreatitis may be avoided by the use of continuous venovenous hemofiltration59 or decompressive anterior abdominal fasciotomy.60 Infection is a common complication of acute necrotizing pancreatitis, and its development is associated with an increased risk of morbidity and mortality.61 For the patient with severe acute pancreatitis, these infections may arise within the injured or necrotic peripancreatic tissues or at distant sites as nosocomial infection in a critically ill patient. The characteristic microbial flora of peripancreatic infection includes organisms normally resident within the gastrointestinal tract62 and organisms that characteristically colonize the proximal gastrointestinal tract of the critically ill patient35 (Box 77.1). Anaerobes may be present but are uncommon, whereas organisms such as Candida, Enterococci, and coagulase-negative Staphylococci are encountered with increasing frequency.63,64 This infecting flora reflects the role of the gastrointestinal tract as the reservoir of organisms inducing superinfection during acute pancreatitis.65
Acute Pancreatitis
Introduction
Definitions and Terminology
Term
Definition
Acute pancreatitis
Acute inflammation of the pancreas with variable involvement of peripancreatic and remote tissues
Mild acute pancreatitis
Edema of pancreas; benign clinical course with minimal organ dysfunction and full recovery
Severe acute pancreatitis
Evidence of pancreatic necrosis; complications of infection, pseudocyst; clinical course characterized by organ failure
Acute fluid collection
Pancreatic or peripancreatic fluid, evident early in course of disease, and lacking wall
Pancreatic pseudocyst
Contained collection of pancreatic juice within a capsule of fibrous tissue and arising following an episode of acute pancreatitis
Pancreatic necrosis
Diffuse or focal loss of viability of pancreatic parenchyma or peripancreatic fat, evident as nonenhancing tissue on a contrast-enhanced CT scan
Pancreatic abscess
Localized collection of pus within pancreas or peripancreatic region and containing little or no necrotic material
Pathogenesis
Etiology and Risk Factors
Clinical Presentation and Diagnosis

Grade
Findings
Grade A
Normal pancreas
Grade B
Pancreatic enlargement
Grade C
Pancreatic or peripancreatic inflammation
Grade D
Single peripancreatic fluid collection
Grade E
2 or more pancreatic collections and/or retroperitoneal air
Early Management of the Critically Ill Patient with Acute Pancreatitis
Initial Resuscitation
Infection Prophylaxis