Acute Pain Management
Stephen M. Macres
Peter G. Moore
Scott M. Fishman
Key Points
Related Matter
Nociceptive Pathways
Pain Sensitization
Pain Processing
Universal Pain Assessment Tool
Brachial Plexus
Interscalene Block
Supraclavicular Block
Femoral Nerve Block
Ultrasound Guided Saphenous Nerve Block
Ultrasound Guided Poplietal Nerve Block
TAP Block
Introduction
There are approximately 75 million surgical procedures performed each year in the United States and more than half are performed in the inpatient setting. Appropriate management of acute perioperative pain using multimodal or balanced analgesia is therefore crucial. In 1992 clinical practice guidelines were promulgated by the Agency for Health Care Policy and Research, which provided guidelines for physicians for the treatment of acute pain.1 Soon thereafter the American Society of Anesthesiologists developed treatment guidelines for acute postoperative pain, which have been updated and amended as of 2012.2 Despite significant advances in our knowledge and treatment of acute pain and dissemination of these guidelines, significant deficits continue to persist and the management of acute postoperative pain is still less than optimal.
Acute Pain Defined
Acute pain has been defined as “the normal, predicted, physiologic response to an adverse chemical, thermal, or mechanical stimulus.”5 Generally, acute pain resolves within 1 month. However, poorly managed acute pain that might occur following surgery can produce pathophysiologic processes in both the peripheral and central nervous systems that have the potential to produce chronicity.4 Acute pain-induced change in the central nervous system is known as neuronal plasticity. This can result in sensitization of the nervous system resulting in allodynia and hyperalgesia. Surgical procedures that can be associated with chronic painful conditions include amputation of a limb, lateral thoracotomy, inguinal herniorrhaphy, abdominal hysterectomy, saphenous vein stripping, open cholecystectomy, nephrectomy, and mastectomy.4
Anatomy of Acute Pain
with cell bodies located in the dorsal root ganglia. The first-order neurons that make up the dual ascending system have their origins in the periphery as A delta and polymodal C fibers (Table 56-1). A delta fibers transmit “first pain,” which is described as sharp or stinging in nature and is well localized. Polymodal C fibers transmit “second pain,” which is more diffuse in nature and is associated with the affective and motivational aspects of pain. First-order neurons synapse on second-order neurons in the dorsal horn primarily within laminas I, II, and V, where they release excitatory amino acids and neuropeptides (Figs. 56-3 and 56-4). Some fibers can ascend or descend in Lissauer’s tract prior to terminating on neurons that project to higher centers. Second-order neurons consist of nociceptive-specific and wide dynamic-range (WDR) neurons. Nociceptive-specific neurons are located primarily in lamina I, respond only to noxious stimuli, and are thought to be involved in the sensory-discriminative aspects of pain. WDR neurons are predominately located in laminae IV, V, and VI, respond to both nonnoxious and noxious input, and are involved with the affective–motivational component of pain. Axons of both nociceptive-specific and WDR neurons ascend the spinal cord via the dorsal column-medial lemniscus and the anterior lateral spinothalamic tract to synapse on third-order neurons in the contralateral thalamus, which then project to the somatosensory cortex where nociceptive input is perceived as pain (Fig. 56-1).
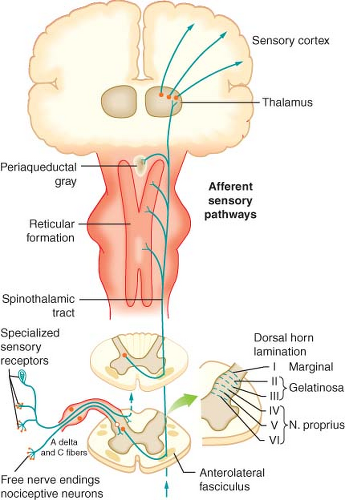 Figure 56.1. Afferent nociceptive pathway. |
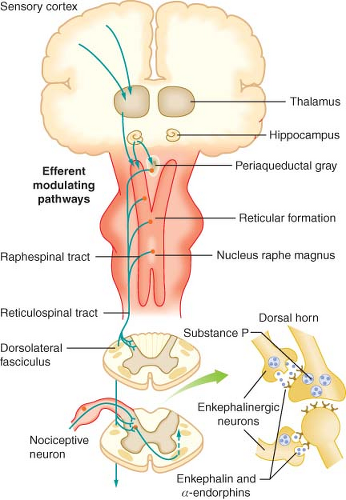 Figure 56.2. Efferent pathways involved in nociceptive regulation. |
Table 56-1. Primary Afferent Nerves | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
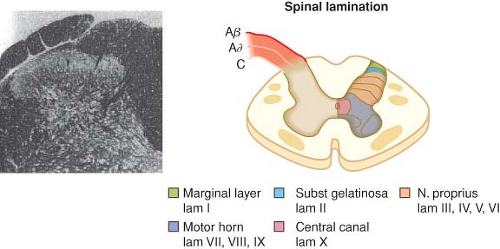 Figure 56.3. Schematic on the right showing the Rexed lamination and the approximate organization of the approach of the afferent to the spinal cord as they enter at the dorsal root entry zone and then penetrate into the dorsal horn to terminate in laminae I and II (A/C) or penetrate more deeply to loop upward to terminate as high as the dorsum of lamina III (Aβ). Inset on the left shows histologic appearance of the left dorsal quadrant, and large, myelinated axons. (From: Warfield CA, Bajwa ZH, eds. Principles and Practice of Pain Medicine. 2nd ed. New York, NY: McGraw Hill Medical Publishing Division; 2004.) |
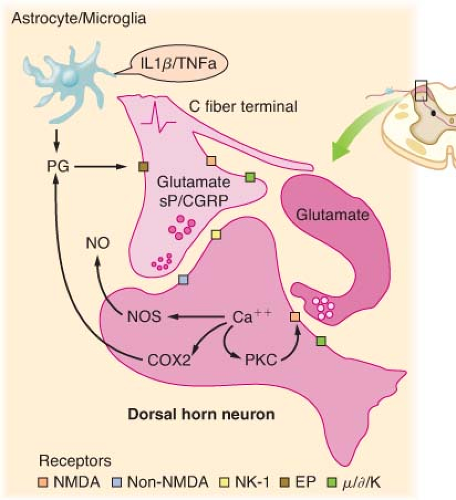 Figure 56.4. Schematic summarizing the organization of dorsal horn systems that contribute to the processing of nociceptive information. (1) Primary afferent C fibers release peptide (e.g., substance P [sP], calcitonin gene–related peptide [CGRP], and so on) and excitatory amino acid (glutamate) products. Small dorsal root ganglion (DRG) cells, as well as some postsynaptic elements contain nitric oxide synthase (NOS) and are able, upon depolarization, to release NO (nitric oxide). (2) Peptides and excitatory amino acids evoke excitation in second-order neurons. For glutamate, direct monosynaptic excitation is mediated by non–N-methyl-D-aspartate (NMDA) receptors (i.e., acute primary afferent excitation of WDR neurons is not mediated by the NMDA or neurokinin 1 (NK-1) receptor). (3) Interneurons excited by afferent barrage induce excitation in second-order neurons via an NMDA receptor. This leads to a marked increase in intracellular Ca2+ and the activation of kinases and phosphorylating enzymes. Prostaglandins (PGs) generated by cyclooxygenase-2 (COX-2) and NO by NOS are formed and released. These agents diffuse extracellulary and facilitate transmitter release (retrograde transmission) from primary and nonprimary afferent terminals, either by a direct cellular action (e.g., NO) or by an interaction with a specific class of receptors (e.g., EP receptors for prostanoids). (4) Nonneuronal sources of prostaglandins may include activated astrocytes and microglia that are stimulated by circulating cytokines, which are released secondary to peripheral nerve injury and inflammation. Terminal excitability can be altered by activation of a variety of receptors located on the sensory terminal, including those for, μ, δ, and κ opioids. (From: Warfield CA, Bajwa ZH, eds. Principles and Practice of Pain Medicine. 2nd ed. New York, NY: McGraw Hill Medical Publishing Division; 2004.) |
Pain Processing
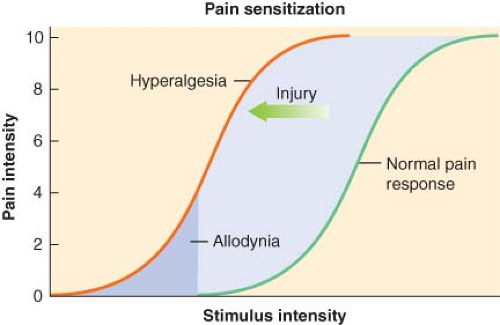 Figure 56.5. Pain sensitization. (Obtained with permission from: Dave Klemm, http:www.georgetown.edu/dml/facs/graphics/index.htm.) |
Chemical Mediators of Transduction and Transmission
Tissue damage following surgical procedures leads to the activation of small nociceptive nerve endings and local inflammatory cells (e.g., macrophages, mast cells, lymphocytes, and platelets) in the periphery. Antidromic release of substance P and glutamate from small nociceptive afferents results in vasodilation, extravasation of plasma proteins, and stimulation of inflammatory cells to release numerous algogenic substances (Table 56-2 and Fig. 56-7). This chemical milieu will both directly produce pain transduction via nociceptor stimulation as well as facilitate pain transduction
by increasing the excitability of nociceptors. Peripheral sensitization of polymodal C fibers and high-threshold mechanoreceptors by these chemicals leads to primary hyperalgesia, which by definition is an exaggerated response to pain at the site of injury.
by increasing the excitability of nociceptors. Peripheral sensitization of polymodal C fibers and high-threshold mechanoreceptors by these chemicals leads to primary hyperalgesia, which by definition is an exaggerated response to pain at the site of injury.
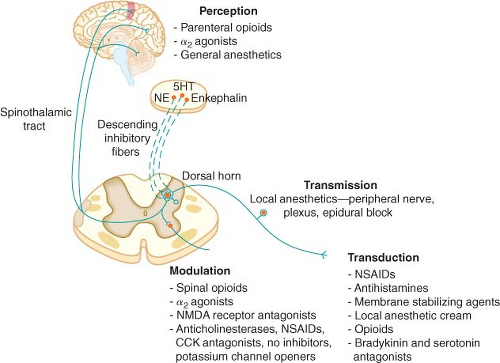 Figure 56.6. The four elements of pain processing: Transduction, transmission, modulation, and perception. 5HT, serotonin; NE, norepinephrine; NMDA, N-methyl-D-asparate; NSAIDs, nonsteroidal anti-inflammatory drugs; CCK, cholecystokin; NO, nitric oxide. |
As is the case in the periphery, the dorsal horn of the spinal cord contains numerous transmitters and receptors involved in pain processing. Three classes of transmitter compounds integral to pain transmission include (1) the excitatory amino acids glutamate and aspartate, (2) the excitatory neuropeptides substance P and neurokinin A, and (3) the inhibitory amino acids glycine and GABA. The various pain receptors include (1) the NMDA (N-methyl-D-aspartate), (2) the AMPA (α-amino-3-hydroxy-5-methylisoxazole-4-proprionic acid), (3) the kainate, and (4) the metabotropic (Fig. 56-8).
Table 56-2. Algogenic Substances | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
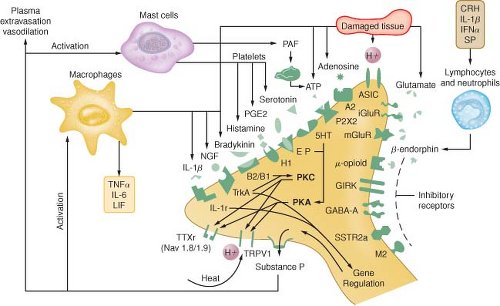 Figure 56.7. Schematic of the neurochemistry of somatosensory processing at peripheral sensory nerve endings. (From: Benzon HT, Raja SN, Molloy RE, et al., eds. Essentials of Pain Medicine and Regional Anesthesia. 2nd ed. Elsevier, Churchill Livingstone; 2005:8.) |
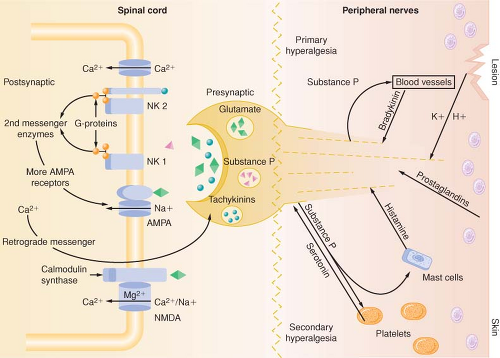 Figure 56.8. Schematic representation of peripheral and spinal mechanism involved in neuroplasticity. Primary hyperalgesia results from tissue release of toxic substances. These toxic substances spread to adjacent tissues, prolonging the hyperalgesic state (secondary hyperalgesia). As C fiber terminals increase in frequency of release of neurotransmitters, such as glutamate, substance P, tachykinins, brain-derived neurotrophic factor, and calcitonin gene–related peptide, the effects of these neurotransmitters are summated, resulting in prolonged depolarizations of second-order neurons (“wind-up”). Function changes at the second-order neuron occur as a result of neurotransmitter binding to postsynaptic receptors, which results in activity-dependent plasticity of the spinal cord. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; NK, neurokinin; NMDA, N-methyl-D-asparate. |
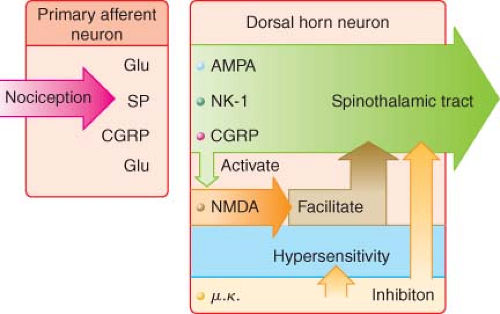 Figure 56.9. Primary nociceptive transmission in the spinal cord. Primary afferent nociceptive input is transmitted via α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), neurokinin-1 (NK1), and calcitonin gene–related peptide (CGRP) synapses, whose signals work their way to the thalamus. Glutaminergic (N-methyl-D-asparate [NMDA]) synapses do not participate significantly in primary nociceptive transmission, but instead play a crucial role in spinal sensitization. Accordingly, even after complete NMDA blockade in the spinal cord, primary afferent nociceptive information is transmitted to the thalamus. NMDA antagonists thus have an antihyperalgesic rather than an analgesic effect in the spinal cord. Glu, glutamate; SP, substance P. (Reprinted from: International Association for the Study of Pain: Pain Control Updates. IASP Newletter 2005;13(2):3, with permission.) |
The AMPA and kainate receptors, which are sodium channel dependent, are essential for fast synaptic afferent input. On the other hand, the NMDA receptor, which is calcium channel dependent, is only activated following prolonged depolarization of the cell membrane. Release of substance P into the spinal cord will remove the magnesium block on the channel of the NMDA receptor giving glutamate free access to the NMDA receptor. Repetitive C-fiber stimulation of WDR neurons in the dorsal horn at intervals of 0.5 to 1 Hz can precipitate the occurrence of wind-up and central sensitization (Fig. 56-9). This leads to secondary hyperalgesia, which, by definition, is an increased pain response evoked by stimuli outside the area of injury.
The Surgical Stress Response
Although similar, postoperative pain and the surgical stress response are not the same. Surgical stress causes release of cytokines (e.g., interleukin-1, interleukin-6, and tumor necrosis factor-α) and precipitates adverse neuroendocrine and sympathoadrenal responses, resulting in detrimental physiologic responses, particularly in high-risk patients.4
The increased secretion of the catabolic hormones such as cortisol, glucagon, growth hormone, and catecholamines and the decreased secretion of the anabolic hormones such as insulin and testosterone, characterize the neuroendocrine response. The end result of this is hyperglycemia and a negative nitrogen balance, the consequences of which include poor wound healing, muscle wasting, fatigue, and impaired immunocompetency.
The sympathoadrenal response has detrimental effects on numerous organ systems; these are listed in Table 56-3.8
Table 56-3. Consequences of Poorly Managed Acute Pain | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Preventive Analgesia
Strategies for Acute Pain Management
The majority of postoperative pain is nociceptive in character, but there are a small percentage of patients who can experience neuropathic pain postoperatively. It is critical to recognize this
fact because patients with neuropathic pain are at increased risk of progressing to a chronic pain state. Neuropathic pain is a result of accidental nerve injury secondary to cutting, traction compression, or entrapment.4 Clinical features may include continuous burning, paroxysmal shooting, or electric pain with associated allodynia, hyperalgesia, and dysesthesias. There can be a delay in the onset of the pain, and it can follow a nondermatomal distribution. Surgical procedures that are a relatively high risk for neuropathic pain include limb amputations, breast surgery, gallbladder surgery, thoracic surgery, and inguinal hernia repair.4 Nociceptive pain responds best to opioids, nonsteroidal anti-inflammatory drugs (NSAIDs), para-aminophenol agents, and regional anesthesia techniques.10 Neuropathic pain, on the other hand, may benefit from the addition of the nonopioid analgesic adjuvants such as the NMDA receptor antagonists, α2-agonists, and the α2–δ subunit calcium channel ligands, which will be discussed in detail.
fact because patients with neuropathic pain are at increased risk of progressing to a chronic pain state. Neuropathic pain is a result of accidental nerve injury secondary to cutting, traction compression, or entrapment.4 Clinical features may include continuous burning, paroxysmal shooting, or electric pain with associated allodynia, hyperalgesia, and dysesthesias. There can be a delay in the onset of the pain, and it can follow a nondermatomal distribution. Surgical procedures that are a relatively high risk for neuropathic pain include limb amputations, breast surgery, gallbladder surgery, thoracic surgery, and inguinal hernia repair.4 Nociceptive pain responds best to opioids, nonsteroidal anti-inflammatory drugs (NSAIDs), para-aminophenol agents, and regional anesthesia techniques.10 Neuropathic pain, on the other hand, may benefit from the addition of the nonopioid analgesic adjuvants such as the NMDA receptor antagonists, α2-agonists, and the α2–δ subunit calcium channel ligands, which will be discussed in detail.
Strategies for acute pain management should also consider the sex of the patient as sex differences appear to exist for pain perception as well as response to opioid analgesics. Evidence suggests that women experience more pain following surgery than men, and therefore require more morphine to achieve a similar level of pain relief.11
Assessment of Acute Pain
A variety of well-studied pain measurement scales exist that can be helpful yet are not definitive. Unidimensional instruments such as the familiar numerical pain scale already mentioned, the visual analog scale, and the “faces” (Fig. 56-10) pain rating scale can provide some degree of guidance about a patient’s experience of pain, but all of these are completely subjective and are open to wide variation between subjects and within subjects at different times.
Multidimensional instruments, such as the McGill Pain Questionnaire or the Brief Pain Inventory provide a broader picture of a patient’s experience, but are usually more cumbersome to administer and, in the end, suffer the same limitations as all other attempts to measure pain. A number of tools to assess cancer-related and noncancer chronic pain have been advanced and validated.12 Most of these focus on persistent background pain and do not help identify intermittent or breakthrough pain. Several assessment scales specifically address breakthrough or episodic pain. The Breakthrough Pain Questionnaire was introduced by Portenoy and Hagen to assess breakthrough pain in cancer
patients, and has also been studied in patients with acute noncancer pain, for which it can offer a picture of both breakthrough and background pain states.13
patients, and has also been studied in patients with acute noncancer pain, for which it can offer a picture of both breakthrough and background pain states.13
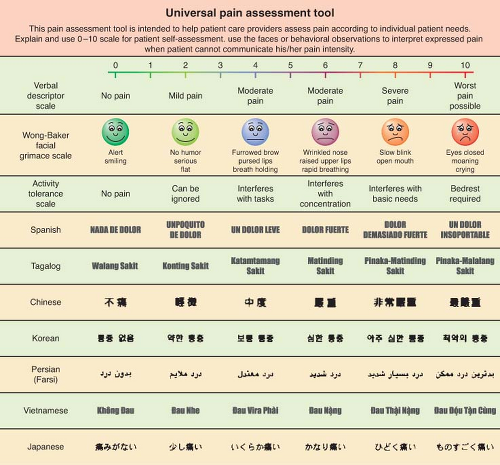 Figure 56.10. Linear verbal analog score and faces pain assessment tool. |
Table 56-4. Three Classes of Acute Pain | |
|---|---|
|
Ultimately, we are left with a maxim first attributed to Dr. John Bonica, the father of pain medicine: “Pain is what a patient says it is.” The best way to begin assessing a patient’s pain is to ask about it and listen to the answers. Attempts to reduce the experience to finite details may lead to failure to ask the right questions, distance us from our patients, focus us away from the whole person, and potentially miss golden diagnostic clues that could lead to effective interventions.
Effective treatment of acute pain requires assessment as well as vigilant reassessment to determine if the primary goals are met, adversity has occurred, or changes are necessary. Acute pain may be viewed as breakthrough, intermittent, or background in nature (Table 56-4). The assessment process for each of these is relatively similar and will help to resolve the related condition into broad pathophysiologic groups such as cancer versus noncancer, and nociceptive versus neuropathic, or mixed pain states. Such an approach supports a rational process for developing a useful differential diagnosis and approaches. Table 56-5 lists the common features of pain that are usually reviewed during the assessment for acute pain. A thorough physical examination must also be performed with particular attention to the neurologic examination, which may offer clues to aberrant neural processing. Such neurologic findings may indicate nerve injury, alerting the astute clinician to a neuropathic rather than a nociceptive pain state that requires a different analgesic approach.14 A provocative physical examination may include examination of the affected areas with maneuvers that may provoke pain such as range of motion testing, walking, and cough. The benefits of provocative testing must outweigh the associated suffering incurred by the patient. Medical imaging is also a common part of the acute pain workup. Overemphasis on imaging data should, however, be avoided as this can potentially lead to misinterpretation of the patient’s underlying pain syndrome.
Table 56-5. Features of Pain Commonly Addressed During Assessment | |
|---|---|
|
Opioid Analgesics
There is great diversity in the available routes of administration of opioid analgesics. Table 56-6 is a list of relevant pharmacokinetic data. Table 56-7 offers equianalgesic dosing guidelines for the various opioids. The reader is referred to the section “Perioperative Pain Management of the Opioid-dependent Patient” for a complete discussion of incomplete cross-tolerance between the different opioids and dosing considerations.
Common opioid-induced side effects include sedation, nausea and vomiting, respiratory depression, and constipation. Less common side effects include confusion, urinary retention, dizziness, and myoclonus. Tolerance rarely develops to the constipating effects of the opioids. Alvimopan, a peripherally acting mu receptor antagonist that has negligible systemic absorption, attenuates opioid-induced constipation and shortens postoperative ileus and length of hospital stay.15 Opioid-induced hyperalgesia (OIH) is a relatively rare phenomenon whereby patients who are receiving opioids suddenly and paradoxically become more sensitive to pain despite continued treatment with opioids. Evidence suggests that OIH is more likely to develop following high doses of phenanthrene opioids such as morphine.16 Changing the opioid to a phenyl piperidine derivative such as fentanyl may thwart OIH. There is also evidence that coadministration of an NMDA receptor
antagonist can abolish opioid-induced tolerance and OIH.16 Finally, opioid analgesics have profound immunomodulatory effects, which include inhibition of cellular and humoral immune functions, depressed natural killer cell activity, promotion of angiogenesis and inhibition of apoptosis. Such effects can be beneficial or deleterious depending upon the clinical situation.17,18
antagonist can abolish opioid-induced tolerance and OIH.16 Finally, opioid analgesics have profound immunomodulatory effects, which include inhibition of cellular and humoral immune functions, depressed natural killer cell activity, promotion of angiogenesis and inhibition of apoptosis. Such effects can be beneficial or deleterious depending upon the clinical situation.17,18
Table 56-6. Opioid Analgesic Pharmacokinetics | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 56-7. Opioid Equianalgesic Dosing | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Morphine is the prototype opioid and is the “gold standard” to which all other analgesics are compared. Although the plasma half-life of the drug is approximately 2 hours its analgesic duration of action is closer to 4 to 5 hours. Morphine undergoes hepatic glucuronidation to morphine-6-glucuronide and morphine-3-glucuronide, both of which are cleared by the kidney. Morphine-6-glucuronide is an active metabolite of morphine and is thought to be responsible for most of the analgesia associated with chronic dosing of the drug. Morphine-3-glucuronide, on the other hand, is considered to be devoid of analgesic activity. With chronic dosing these metabolites can accumulate and can be particularly problematic in patients with renal failure. Dosing adjustment is therefore necessary and monitoring of side effects is important. Morphine-6-glucuronide contributes to side effects such as drowsiness, nausea and vomiting, coma, and respiratory depression. Morphine-3-glucuronide, on the other hand, is thought to cause agitation, myoclonus, delirium, and hyperalgesia.
Hydromorphone is a semisynthetic opioid that has 4 to 6 times the potency of morphine. It is available for oral, rectal, parenteral, and neuraxial administration. Whereas the oral bioavailability of the drug is reported to be 20% to 50%, its bioavailability via the subcutaneous route is 78%, making it the ideal drug for long-term subcutaneous administration in the opioid-tolerant patient. Like morphine, hydromorphone is biotransformed in the liver. The active metabolites are dihydromorphine and dihydroisomorphine and the inactive metabolite is hydromorphone-3-glucuronide. Although hydromorphone has traditionally been the preferred opioid for patients with acute pain and impaired kidney function, evidence suggests that hydromorphone-3-glucuronide can accumulate in those with renal failure and may contribute to side effects such as neuroexcitation and cognitive impairment. Opioid-related side effects such as nausea, vomiting, sedation, cognitive impairment, and pruritus are reported to be less intense with hydromorphone vis-à-vis morphine. In fact, the incidence of pruritus following neuraxial administration of hydromorphone is reported to be approximately 5% versus the 11% to 77% range reported for neuraxial morphine.19
Fentanyl, a synthetic opioid chemically related to the phenylpiperidines, is a relatively selective mu receptor agonist, which is considered to be 80 times the potency of morphine following intravenous administration. It is extensively metabolized in the liver to norfentanyl and other inactive metabolites, which are excreted in the urine and bile. Fentanyl is therefore suitable for patients in renal failure. The drug is available for intravenous, subcutaneous, transdermal, transmucosal, and neuraxial administration. The transdermal administration of fentanyl using iontophoresis (Ionsys, Janssen-Cilag LTD) is a novel on-demand drug delivery system that does not require venous access.
Sufentanil, alfentanil, and remifentanil are analogs of fentanyl that have analgesic effects similar to those of morphine and the other mu receptor agonists. Sufentanil is approximately 1,000 times the potency of morphine and is primarily used in the operating room either intravenously or neuraxially.20 Like fentanyl, sufentanil is very lipophilic, and although their pharmacokinetic and pharmacodynamic profiles are similar, sufentanil has a smaller volume of distribution and shorter elimination half-life.20 The high intrinsic potency of sufentanil makes it an excellent choice for epidural analgesia in the opioid-dependent patient.21 Alfentanil is approximately 10 times the potency of morphine and, like sufentanil, is used primarily in the operating room either intravenously or neuraxially. Remifentanil is an ultra–short-acting synthetic opioid. The potency of the drug is approximately equal to that of fentanyl. Remifentanil is rapidly degraded by tissue and plasma esterases, which accounts for its incredibly short terminal elimination half-life of 10 to 20 minutes.20 Rapid clearance and lack of accumulation make this a very desirable opioid in the operative setting. One disadvantage, however, is that discontinuation of a remifentanil infusion results in rapid loss of analgesia. There is also evidence to suggest that remifentanil infusions may be associated with the development of opioid-induced hyperalgesia. Further studies are clearly needed to better define this phenomenon.
Meperidine, a phenylpiperidine, is a synthetic mu opioid receptor agonist with a short half-life. The drug is recommended for the short-term management of acute pain only and has absolutely no role in the management of chronic pain. The drug is biotransformed by the liver to normeperidine, a potentially neurotoxic metabolite, which has a 12- to 16-hour half-life. Repetitive dosing of meperidine can cause accumulation of normeperidine, which may precipitate tremulousness, myoclonus, and seizures. It is therefore recommended that the total daily intravenous dose in an otherwise healthy adult without renal or central nervous system disease should not exceed 600 mg/day and should not be administered for longer than 48 hours.22 We do not recommend administration of meperidine as an intravenous patient–controlled analgesia (PCA). The drug is contraindicated in patients receiving monoamine oxidase inhibitors as this may precipitate a syndrome characterized by muscle rigidity, hyperpyrexia, and seizures.
Methadone is a relatively inexpensive synthetic opioid considered to be a broad-spectrum opioid because it is a (1) mu receptor agonist, (2) NMDA antagonist, and (3) inhibitor of monoamine transmitter reuptake, making it potentially useful for the treatment
of neuropathic pain. The drug is well absorbed from the gastrointestinal tract with a reported bioavailability of approximating 80%. The drug is extensively metabolized in the liver by the cytochrome P450 (CYP450) system to inactive metabolites, which are cleared in the bile and urine; unlike morphine, it is generally not necessary to adjust the dosage of methadone in patients with renal insufficiency. Methadone has an elimination half-life of 22 hours, and following a single dose the duration of analgesia is approximately 3 to 6 hours. With repetitive dosing, however, methadone can accumulate and slow tissue release into the blood stream can result in a long elimination half-life of up to 128 hours and duration of analgesia of 8 to 12 hours. This long half-life explains the potential risk for cumulative toxicity, and therefore the importance of monitoring for side effects such as excessive sedation and confusion following the initiation of an around-the-clock dosing regimen.
of neuropathic pain. The drug is well absorbed from the gastrointestinal tract with a reported bioavailability of approximating 80%. The drug is extensively metabolized in the liver by the cytochrome P450 (CYP450) system to inactive metabolites, which are cleared in the bile and urine; unlike morphine, it is generally not necessary to adjust the dosage of methadone in patients with renal insufficiency. Methadone has an elimination half-life of 22 hours, and following a single dose the duration of analgesia is approximately 3 to 6 hours. With repetitive dosing, however, methadone can accumulate and slow tissue release into the blood stream can result in a long elimination half-life of up to 128 hours and duration of analgesia of 8 to 12 hours. This long half-life explains the potential risk for cumulative toxicity, and therefore the importance of monitoring for side effects such as excessive sedation and confusion following the initiation of an around-the-clock dosing regimen.
Table 56-8. Conversion Ratios from Morphine to Methadone | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Finally, opioid rotation is a very useful technique to restore analgesic sensitivity in the highly tolerant patient, and methadone is a common choice for opioid rotation. Because cross-tolerance is incomplete, the calculated equianalgesic dose of any new opioid is always lower than expected. One must be particularly cautious, however, when converting from morphine to methadone as the morphine/methadone equianalgesic ratio appears to be curvilinear; whereas the morphine-to-methadone conversion ratio is 3:1 at morphine doses of <100 mg/day, the ratio is 20:1 at morphine doses of >1,001 mg/day (Table 56-8). Methadone is principally metabolized by the CYP3A4 subtype enzyme of the cytochrome P450 system and to a lesser extent by the CYP1A2 and CYP2D6 subtypes. Consequently, there is the potential for numerous drug interactions with methadone, as shown in Table 56-9. Whereas inhibition of methadone metabolism will theoretically provoke
toxicity, induction of methadone metabolism could potentially precipitate inadequate analgesia or even withdrawal symptoms. Frequent adjustments of the methadone dosage may therefore be required if medications are added to or eliminated from a patient’s drug regimen. A rare side effect associated with methadone is a pause-dependent dysrhythmia associated with bradycardia, QT prolongation, and Torsades de point.
toxicity, induction of methadone metabolism could potentially precipitate inadequate analgesia or even withdrawal symptoms. Frequent adjustments of the methadone dosage may therefore be required if medications are added to or eliminated from a patient’s drug regimen. A rare side effect associated with methadone is a pause-dependent dysrhythmia associated with bradycardia, QT prolongation, and Torsades de point.
Table 56-9. Methadone Drug Interactions | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
Buprenorphine is a potent semisynthetic opioid that is derived from thebaine. The drug is classified as a mixed agonist–antagonist and partial mu receptor agonist with an analgesic potency 25 to 50 times greater than that of morphine. Buprenorphine is a lipophilic opioid with moderate intrinsic activity and a high affinity for the mu opioid receptor, with a half-life for dissociation from the receptor of 166 minutes compared with 7 minutes for fentanyl. The drug is metabolized by the gut and the liver and has a half-life of about 3 hours, but this bears little connection to the rate of disappearance of its clinical effects because of its avid binding to the mu receptor, as previously noted. The drug can be delivered by various routes of administration include intravenous, intramuscular, neuraxial, subcutaneous, sublingual, and transdermal. Buprenorphine is also an excellent alternative for the treatment of acute pain in the patient who cannot tolerate morphine secondary to allergy or other sensitivity. In humans, buprenorphine is reported to have a ceiling effect for respiratory depression but not for analgesia. Buprenorphine is reported to have anti-inflammatory effects and therefore may be efficacious when administered intra-articularly. Investigators have demonstrated that buprenorphine will significantly prolong the analgesic effects of a peripheral nerve block when 0.3 mg of the drug is combined with 40 mL of a local anesthetic mixture consisting of 1% mepivacaine, 0.2% tetracaine, and epinephrine 1:200,000.23
Nonopioid Analgesic Adjuncts
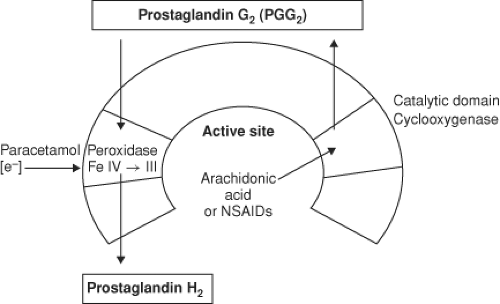 Figure 56.11. From Kam, P.C.A., COX-3: Uncertainties and controversies, Curr Anaesth Crit Care. 2009; (20) 50. |
COX-1 is the constitutive enzyme that produces prostaglandins, which are important for general “house-keeping” functions such as gastric protection and hemostasis. COX-2, on the other hand, is the inducible form of the enzyme that produces prostaglandins that mediate pain, inflammation, fever, and carcinogenesis. Prostaglandin E2 is the key mediator of both peripheral and central pain sensitization. Peripherally, prostaglandins do not directly mediate pain; rather, they contribute to hyperalgesia by sensitizing nociceptors to other mediators of pain sensation such as histamine and bradykinin.24 Centrally, prostaglandins enhance pain transmission at the level of the dorsal horn by (1) increasing the release of substance P and glutamate from first-order pain neurons, (2) increasing the sensitivity of second-order pain neurons, and (3) inhibiting the release of neurotransmitters from the descending pain-modulating pathways.
NSAIDs have proved effective in the treatment of postoperative pain. In addition, they are opioid-sparing and can significantly decrease the incidence of opioid-related side effects such as postoperative nausea and vomiting and sedation.25 Unlike the opioids, NSAIDs exhibit a “ceiling effect” with respect to maximum analgesic effects. Parenteral NSAIDs such as ketorolac are commonly employed as part of a multimodal approach for acute perioperative pain management. The optimal dose of ketorolac for postoperative pain control is 15 to 30 mg intravenously every 6 to 8 hours not to exceed 5 days. The dose should be decreased in patients with renal failure.
Despite the benefits of NSAIDs in the perioperative period they are not without some significant side effects. Platelet dysfunction, gastrointestinal ulceration, and an increased risk of nephrotoxicity are several reasons why the nonselective NSAIDs may be avoided in the perioperative period. The risk of nephrotoxicity is increased in patients with hypovolemia, congestive heart failure, and chronic renal insufficiency.24 The COX-2–selective inhibitors were developed in an attempt to minimizing their side effects. The COX-2–specific inhibitor celecoxib (Celebrex) is available in the United States. Rofecoxib (Vioxx) and valdecoxib (Bextra) also released in the same period were recalled by the manufacturers because of concerns about adverse cardiovascular risks. Celecoxib is the only COX-2–specific inhibitor currently available in the United States for acute postoperative pain. The recommended oral loading dose is a 400 mg followed by 200 mg orally every 12 hours for several days. Parecoxib (Dynastat) is an injectable COX-2–specific inhibitor that is available only in Europe for the treatment of moderate-to-severe postoperative pain. The recommended dose of the drug in Europe is 40 mg intravenously or intramuscularly initially followed by 20 to 40 mg every 4 to 6 hours not to exceed 80 mg/day. Unlike the nonselective NSAIDs, however, COX-2–specific inhibitors offer the potential advantages of a reduced incidence of gastrointestinal ulceration and they do not inhibit platelet function. Because prostaglandins play a crucial role in renal function through their effect on blood flow, natriuresis and glomerular filtration, traditional NSAIDs, and the COX-2 inhibitors can cause fluid retention and hypertension.
The authors, therefore, do not recommend prescribing a COX-2 inhibitor for patients with a known history of coronary artery disease or cerebrovascular disease. Both COX-1 and COX-2 play significant roles in bone fusion following fracture, and the use of the traditional NSAIDs has been found to inhibit the healing process, particularly following lumbar spinal fusion surgery. The effect of COX-2 inhibitors on bone fusion following orthopedic procedures continues to be controversial, and no recommendations can be made at this time. NSAIDs and
COX-2–selective inhibitors should not be administered to patients with known hypersensitivity to the drugs or to patients with Samters triad (aka aspirin triad), which is a medical condition consisting of asthma, aspirin insensitivity, and nasal polyposis. Finally, avoid celecoxib and valdecoxib in patients with allergic-type reactions to sulfonamides.
COX-2–selective inhibitors should not be administered to patients with known hypersensitivity to the drugs or to patients with Samters triad (aka aspirin triad), which is a medical condition consisting of asthma, aspirin insensitivity, and nasal polyposis. Finally, avoid celecoxib and valdecoxib in patients with allergic-type reactions to sulfonamides.
Table 56-10. Nonopioid Analgesics (Adult Dosing Guidelines) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The para-aminophenol derivative acetaminophen (aka paracetamol) has both analgesic and antipyretic properties, similar to aspirin, but is devoid of any anti-inflammatory effects. The drug is primarily a centrally acting inhibitor of the cyclooxygenase enzyme with minimal peripheral effects. Acetaminophen neither enters the active site of the COX enzyme nor binds to the cyclooxygenase site, but instead it prevents COX activation by reducing heme at the peroxidase site of the enzyme (Fig. 56-11). In addition, there may be modulation of descending inhibitory serotoninergic pathways and the drug may act on the opioid, cannabinoid, and NMDA receptors.27 Acetaminophen is devoid of many of the side effects generally associated with the NSAIDs, such as gastrointestinal ulceration, impaired platelet function, adverse cardiorenal effects, and impairment of bone fusion following orthopedic procedures. Acetaminophen is opioid-sparing and can be used in conjunction with an NSAID as part of a multimodal analgesic program. In adults, 2 g of oral acetaminophen is equivalent to 200 mg of celecoxib.
Propacetamol is the intravenous prodrug formulation of paracetamol and is a popular adjuvant for perioperative pain control in Europe. The drug has the disadvantage that it must be reconstituted prior to administration. Two grams of propacetamol is equivalent to 1 g of paracetamol. Fortunately, a ready-to-use intravenous formulation of paracetamol (Perfalgan) has been released in Europe. Intravenous acetaminophen (Ofirmev) was released in the United States in November of 2010. The drug is available as a 1 g (1,000 mg/100 mL) infusion that does not require reconstitution and can be infused through a peripheral IV over 15 minutes. See Table 56-10 for dosing guidelines.
Related posts:




Full access? Get Clinical Tree


