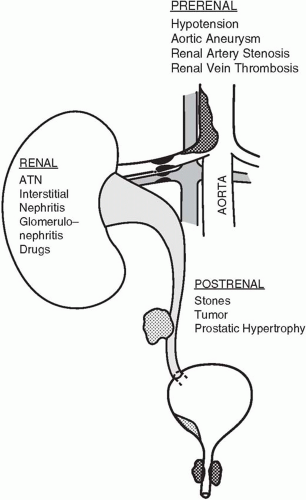
Hypoperfusion accounts for half of all cases of AKI; hence, prevention of hypotension or its rapid reversal is probably the most effective therapy. Although prolonged ischemia alone may lead to AKI, in most cases the etiology is multifactorial (i.e., severe sepsis, drugs, and hypotension). In general, mean arterial pressures less than 60 to 70 mm Hg for longer than 30 min risk injury, particularly if there is accompanying hypoxia. Hypoperfusion results from circulatory failure, hypotension, vascular obstruction (e.g., renal artery stenosis, vasculitis, embolization of bland clot or cholesterol or maldistribution of cardiac output (CO), as in severe sepsis. The hepatorenal syndrome (see
Chapter 31) and the renal response to positive end-expiratory pressure (PEEP) mimic prerenal physiology by redistributing blood flow away from the filtering glomerulus.
Marked increases in intra-abdominal pressure (abdominal compartment syndrome), usually greater than 25 mm Hg, resulting from the accumulation of ascitic fluid or blood, can also produce a prerenal state. It is uncertain if elevated abdominal pressures lessen urine flow by reducing CO through impaired venous return; by obstructing kidney arterial supply or venous outflow; or by compressing the kidney parenchyma directly. It appears unlikely that ureteral obstruction is etiologic because placement of ureteral stents does not improve flow. Regardless of mechanism, when elevated pressures are responsible for oliguria, decompressive laparotomy or paracentesis is associated with a near immediate restoration of urine flow.
Indicators of Intravascular Volume Status
Orthostatic blood pressure is a helpful clinical measure of intravascular volume status in healthy persons. However, few critically ill patients can be tested in this fashion, and autonomic insufficiency renders such changes less reliable in patients who are diabetic, elderly, or bedridden. Dry mucous membranes, skin laxity, and absence of axillary moisture may also be clues to hypovolemia. Unfortunately, these signs, too, prove unreliable in patients with hyperpnea or advanced age. For the hospitalized patient, a persistently negative fluid balance supports a prerenal diagnosis.
Response to fluid challenge is the diagnostic hallmark of prerenal disease. The rate and volume of fluid administered must be customized taking into account the estimated magnitude of the deficit, and cardiopulmonary reserves. Repeated boluses of 15 to 20 mL/kg are typically administered until deemed futile or function improves. Because noninvasive methods to assess fluid status (chest radiograph and echocardiogram) are insensitive tests of intravascular depletion, invasive monitoring is often undertaken (see
Chapter 2). The need for invasive monitoring is controversial, however, because no particular values for central venous pressure (CVP), pulmonary artery occlusion pressure (PAOP), CO, or systemic vascular resistance (SVR) guarantee sufficient or inadequate renal blood flow. That is,
glomerular pressure and flow are poorly gauged by these hemodynamic measures. Nevertheless, a low intravascular pressure (i.e., CVP ≤2 or PAOP ≤5 mm Hg) can be diagnostically helpful, especially if CO is reduced concomitantly.
In prerenal failure initially the urinalysis is normal or minimally abnormal with hyaline casts and other nonspecific sediment; however, if the injury is sufficient to induce tubular damage, typical ATN sediment will be present. A prerenal state prompts the tubule to reabsorb Na+ and water, yielding concentrated urine with low Na+. A urine Na+ less than 20 mEq/L, urine osmolality greater than 500 mOsm/L, and the urine specific gravity greater than 1.015 are typical. The fractional excretion of sodium (FeNa) is another commonly used index to identify the prerenal state that measures the percentage of filtered sodium that is excreted. FeNa is calculated as the ratio of the urine Na+ concentration times the plasma creatinine concentration to the product of the plasma Na+ concentration times the urine creatinine concentration. FeNa = ([Urine Na+] × [Plasma creatinine])/([Plasma Na+] × [Urine creatinine]) × 100. FeNa values less than 1% are generally indicative of prerenal disease, whereas values greater than 3% usually represent acute tubular necrosis (ATN). Intermediate values are not helpful and there are exceptions to this rule. For example, up to 10% of cases of ATN have a low FeNa. For urine Na+ or FeNa measurements to be valid, the underlying Na+ reabsorbing capacity of the renal tubule must be intact. For this reason, chronic renal failure, hypoaldosteronism, and metabolic alkalosis may all render tests of urine Na+ invalid. Likewise, diuretic therapy invalidates urine Na+ determinations for at least 24 h. Osmotic agents (glucose, mannitol, radiographic contrast) also confuse interpretation of urine chemistry values by diluting the urine.
Although the BUN may be disproportionately high because of increased urea production resulting from tetracycline, corticosteroids, or GI bleeding, an elevated BUN/creatinine ratio more commonly results from reduced tubular urine flow and increased proximal tubular reabsorption of urea nitrogen. Thus, in prerenal states the BUN/creatinine ratio is usually elevated (>10:1; sometimes >20:1). Because the renal tubules cannot absorb creatinine, reduced tubular flow rates have no effect on the creatinine concentration if GFR is preserved. Comparing urea clearance to creatinine clearance will determine whether increased urea production or decreased urea excretion is the cause of an elevated BUN. If the urea/creatinine clearance ratio is greater than 1, increased production is the likely etiology. The most common clinical task is to differentiate prerenal disease from ATN.
Table 29-1 compares the more common renal function indicators in these two conditions. Imaging studies of the kidneys may also provide clues to the cause of renal failure (
Table 29-3). In most forms of AKI, the kidneys will be of normal size when imaged with ultrasound or computed tomography (CT). Small kidneys suggest a more chronic process (e.g., diabetes or hypertension) with a superimposed insult.
Only gold members can continue reading.
Log In or
Register to continue
Related

Full access? Get Clinical Tree