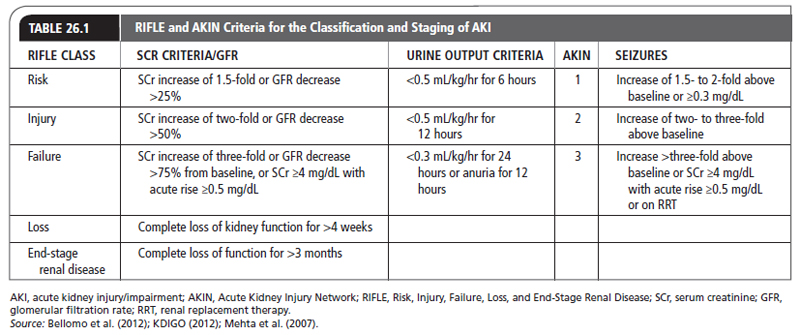
UNIT VI: GENITOURINARY CONDITIONS Karina Muzykovsky, PharmD, BCPS • Kimberly M. Sarosky, MS, PharmD, BCPS Acute kidney injury/impairment (AKI), formerly known as acute renal failure (ARF), is a deterioration of renal function over a period of hours to days that results in failure of the kidney to perform its necessary processes. These include the elimination of nitrogenous waste products, homeostasis of fluids and electrolytes, acid–base balance, gluconeogenesis, and hormone production (erythropoietin, renin, 1,25-dihydroxyvitamin D). An elevation in nitrogenous wastes is referred to as azotemia. Uremia is the clinical manifestation of azotemia and consists of nausea and vomiting, altered sensorium, pruritus, asterixis, decreased appetite, and pericarditis. Previously, more than 30 definitions for ARF existed. The lack of a universally accepted definition was a major limitation for diagnosis and comparison of published literature. In 2004, the Acute Dialysis Quality Initiative (ADQI) group came up with the Risk, Injury, Failure, Loss and End-Stage Renal Disease (RIFLE) criteria for the diagnosis and classification of AKI (Bellomo, Ronco, Kellum, Mehta, & Palevsky, 2004). AKI is broken down into three classes of increasing severity based on changes in serum creatinine (SCr) and urine output; the worst criteria should be used for classification purposes, and two outcome classes are defined by duration of loss of kidney function. In 2007, the Acute Kidney Injury Network (AKIN) slightly modified the RIFLE criteria to indicate that the rise in SCr must occur within 48 hours (Mehta et al., 2007). This shift in terminology reflects the diversity in clinical presentation, from patients with mild renal dysfunction to ones needing renal replacement therapy (RRT). Table 26.1 illustrates these criteria. Both the RIFLE and the AKIN definitions rely upon SCr and urine output. However, the SCr level does not always rise in proportion to renal damage, and there is also a lag period of a day or two between when AKI occurs and when biomarkers become elevated. In the absence of obstruction, urine output directly correlates with the glomerular filtration rate (GFR) in patients with AKI. Anuria is defined as a 24-hour urine production of <50 mL. Oliguria is defined as a 24-hour urine output of 50 to 400 mL. Nonoliguria is defined as a 24-hour urine output of >400 mL. The incidence of AKI ranges from 1% to 67%, depending on the population studied and criteria used to define it (Lameire et al., 2013; Srisawat & Kellum, 2011). AKI is 5 to 10 times more common in the hospital setting than in the community setting and rises to 7.7% to 28.1% in postoperative cardiac surgery patients (Coppolino, Presta, & Saturno, 2013). The highest incidence is seen in critically ill patients with acute tubular necrosis (ATN) in the setting of multiorgan failure. The overall incidence of AKI is increasing owing to an older population with multiple comorbidities (KDIGO, 2012—Suppl Appendix A). However, because of the difficulty in defining and ultimately diagnosing AKI, the exact incidence is unknown. The mortality rate associated with AKI has improved in recent years, probably because of more aggressive nutritional therapy and earlier and improved renal replacement therapies. Before the development of dialytic therapies, the most common causes of death in patients with AKI were uremia, electrolyte abnormalities, acid–base disorders, and volume overload. Fluid overload in particular is associated with an increased risk of death. With the improvement of dialytic techniques, the most common causes of death now are sepsis, cardiovascular (hypotension, arrhythmias) and pulmonary dysfunction, coagulopathy, and the withdrawal of life-support measures. The mortality rate ranges from 15% to 60%, but can be as high as 70% in critically ill patients requiring dialysis (KDIGO, 2012; Lameire et al., 2013). Survival rates for patients with AKI are largely dependent on its original cause and whether other major organ failure is involved. Patients with acute severe parenchymal disease have a worse prognosis than those with ATN, and AKI in the setting of multiorgan failure is an independent risk factor for death. Partial recovery or nonrecovery of renal function represents an independent predictor of long-term mortality for survivors of AKI (Schiffl, 2008). Anatomy and Physiology Normally, people have two kidneys each measuring 10 to 13 cm. Each kidney consists of approximately 1 million nephrons, the functional units of the kidney. The nephrons maintain homeostatic functions by reabsorption of vital constituents (electrolytes, bicarbonate, glucose, essential amino acids) of the blood and secretion and ultimate excretion of what is not necessary (excessive water and electrolytes, medications) as urine. Blood flows to each kidney via the renal artery, which divides into two branches before arriving at the kidneys. Each branch divides into approximately five segmental branches, providing blood flow to their respective portions of the kidney. Each segmental branch further breaks down into smaller arteries and arterioles. Each nephron consists of a glomerulus (a vascular tuft that joins the efferent and afferent arterioles) and the collecting tubules, which consist of the proximal tubule, the loop of Henle, and the distal tubule. In the proximal tubule, 60% to 70% of the filtered load of water and solute is reabsorbed, including amino acids, glucose, and bicarbonate. Reabsorption of calcium, potassium, and magnesium occurs at the loop of Henle, as does maintenance of an osmotic gradient necessary for the concentration of urinary solutes. The distal tubules are responsible for acid–base balance, secretion of potassium, and reabsorption of water. Pathology Risk factors for developing AKI have been defined for different patient populations and are discussed in this chapter. Identification of patients with such risk factors is important in the prevention of AKI. Early detection and intervention are key to minimizing permanent damage to the kidneys. Despite recent advances in the medical care of these patients, the treatment of AKI continues to pose a serious dilemma for the primary care provider. There is an extensive degree of heterogeneity in the development of AKI. Even when a particular insult is known to be present, predicting the likelihood of occurrence is uncertain due to a number of susceptibility factors that vary from patient to patient. The mechanism behind AKI is quite complex, involving a multitude of factors: renal hypoperfusion, volume depletion, enhanced vasoconstriction through autoregulatory pathways, direct insult by exogenous nephrotoxins, proinflammatory mediators, and physical obstruction. Traditionally, AKI is divided into three categories: prerenal (resulting from renal hypoperfusion), intrarenal (resulting from direct damage to the kidney), and postrenal (resulting from a urine flow obstruction). Recently, there has been concern regarding the appropriateness of the use of these anatomical categories (Parikh & Coca, 2010). For example, prerenal AKI has been long associated with intravascular volume depletion or dehydration and the need for rehydration therapy. However, both cardiorenal and hepatorenal syndromes can present in a similar fashion from a laboratory standpoint, but are managed quite differently through fluid restriction (McCullough, Kellum, Mehta, Murray, & Ronco, 2013). Therefore, it is important that the practitioner realize that AKI is a dynamic process, which may or may not involve functional or direct damage to the nephron. Identifying the etiology of AKI involves a differential diagnosis that focuses upon examining both markers of functional status and structural damage, as treatment approaches vary depending upon these factors as well as the individual baseline risk factors of the patient. Noting the multitude of possible underlying causes, only the most commonly encountered examples within clinical practice are addressed in further detail here. For the purpose of simplicity, the three categories of AKI as identified through anatomical location are described herein; however, the preceding caveat should be considered for each individual case. Prerenal AKI results from decreased renal perfusion, with or without systemic hypotension. Depending on a patient’s underlying comorbidities, it is often rapidly reversible with appropriate and timely intervention. Under normal conditions, renal blood flow is approximately 25% of the cardiac output. In states of volume depletion, such as dehydration due to persistent fever or diarrhea, sepsis, or bleeding in the setting of major cardiac or intra-abdominal surgery, there is a decrease in blood volume and consequently a decrease in renal perfusion. Decreased volume status unrelated to blood loss is responsive to administration of intravenous crystalloids such as normal saline (0.9% NaCl) and often reverses this type of AKI. In cases of similar presentation of prerenal AKI, such as of congestive heart failure and cardiogenic shock, there is no decrease in blood volume, but rather a decrease in blood flow to the kidneys, which also results in renal hypoperfusion. Depending on the nature of the cardiac dysfunction and hemodynamic state, intravenous pharmacological measures may be initiated to increase cardiac output with inotropes such as dobutamine, or to decrease preload with nitroglycerin. Through autoregulation, the kidney is capable of maintaining an adequate glomerular and renal blood flow in the presence of moderate reductions in renal perfusion. Prerenal AKI results when autoregulation fails to maintain the GFR. Initiation or dosage increases in medications, which inhibit the renin–angiotensin–aldosterone system (RAAS), such as angiotensin-converting enzyme inhibitors (ACE-I) or angiotensin II receptor blockers (ARBs) are two primary examples of medications that alter these autoregulatory processes. ACE-Is and ARBs act transiently through the inhibition of efferent arteriole vasoconstriction. Additionally, nonsteroidal anti-inflammatory drugs (NSAIDs) affect renal perfusion through a decrease in prostaglandin synthesis, which is required for afferent arteriole vasodilation. If prerenal AKI persists for a prolonged period of time without appropriate management, tissue ischemia may result, with progression to acute intrinsic kidney injury. Acute Intrinsic Kidney Injury Acute intrinsic kidney injury is considered to be the most diverse in terms of underlying cause of injury, as it results from functional or direct structural damage to any part of the kidney, including the small blood vessels, glomerulus, renal tubule, and interstitium. It is important to be able to rule out or distinguish the specific type of acute intrinsic kidney injury through a thorough history and physical examination as well as laboratory and diagnostic studies. The history and physical are important because management strategies may differ. Damage to the small blood vessels may result from cholesterol emboli deposition or malignant hypertension. Damage to the glomerulus can be caused by a number of diseases, including systemic lupus erythematosus and Goodpasture’s syndrome. Injury to the renal tubules may result from severe renal hypoperfusion or direct toxicity from exogenous and endogenous substances. Commonly encountered examples of exogenous nephrotoxic substances include radiocontrast dyes, aminoglycosides, and colistin. Contrast-induced acute kidney injury (CI-AKI) is relatively common in both the inpatient and outpatient settings. KDIGO defines CI-AKI as a rise in SCr ≥0.5 mg/dL, or a 25% increase from baseline approximately 48 hours after radiological procedure (KDIGO, 2012). All contrast dyes are hyperosmolar to that of plasma and most contain iodine, which itself is cytotoxic. Additionally, contrast dyes are vasoconstrictive and highly viscous. They cause a decrease in renal blood flow and increase contact time, causing ischemic nephropathy specifically within the medullary tubules (Seeliger, Sendeski, Rihal, & Persson, 2012). Myoglobin, a potential endogenous nephrotoxin, is released in rhabdomyolysis and may cause acute damage to the renal tubules. Nonselective backleak of filtrate across damaged renal tubules may also result in ATN. Damage to the interstitium may result from infections, such as cytomegalovirus and streptococci; and from many drugs, including penicillin and cephalosporin antibiotics, ciprofloxacin, sulfonamides, phenytoin, and NSAIDs. In the case of acute interstitial nephritis (AIN), presentation may include fever, rashes, eosinophilia, and eosinophiluria. However, eosinophils are usually absent with chronic use of NSAIDs, the most common etiology of drug-induced AIN (Praga & González, 2010). This may be attributed to masking of the inflammatory response. Postrenal Obstruction Postrenal obstruction is characterized by an acute onset of anuria. The incidence of postrenal failure is low: It accounts for <9% of all cases of AKI, which is specifically prominent within the elderly patient (Yilmaz & Erdem, 2010). It must occur in both kidneys to cause AKI, or in one kidney if the patient has a solitary kidney. Crystal deposition from medications such as acyclovir or sulfonamides, and renal caliculi—most commonly as calcium or oxalate—may cause a urinary obstruction. Malignancy, atonic bladder, benign prostatic hypertrophy, or urethral stricture are other conditions that may precipitate obstruction, especially in the presence of anticholinergic medications or an improperly placed urinary catheter. Tables 26.2 and 26.3 outline the most common causes of AKI. A detailed history of present illness is crucial in the initial evaluation in order to identify any underlying risk factors for AKI. A history of concomitant disease states, family medical history, medication use, and illicit substance use or exposure to environmental toxins are all pertinent to the diagnosis. With regard to medication use, examination of all newly initiated medications and dosage increases should be accounted for as well as use of nonprescription medications and herbal supplements. A recent history of an exacerbation of congestive heart failure, volume depletion secondary to fever during prolonged infection, a traumatic injury, or procedures requiring radiocontrast dye may suggest the diagnosis of AKI. A thorough physical examination is a necessary part in the workup of AKI. Clinical findings of thirst and orthostatic dizziness as well as physical findings of orthostatic hypotension, tachycardia, and dry skin turgor suggest prerenal failure secondary to volume depletion. A rash may suggest allergic interstitial nephritis. Flank pain, although predominantly associated with potential infection, may indicate a renal artery or vein occlusion. Digital ischemia may suggest atheroembolization. Gastrointestinal symptoms of nausea and vomiting may also be encountered due to a buildup of uremic toxins. Subsequently, the buildup of uremic toxins may also lead to neurological symptoms of altered mental status. Causes of Acute Kidney Injury PRERENAL POSTRENAL Decreased intravascular volume Bleeding Dehydration Decreased renal blood flow Congestive heart failure Cardiogenic shock Sepsis Vasodilating drugs Prostatic hypertrophy Tumor Stones Emboli Crystals INTRINSIC Glomerular IgA nephropathy Systemic lupus erythematosus Goodpasture’s syndrome Postinfectious Tubulointerstitial Acute tubular necrosis Acute interstitial nephritis Multiple myeloma Pyelonephritis Vascular Wegener’s granulomatosis Polyarteritis nodosa Thrombotic thrombocytopenic purpura Pre-eclampsia Scleroderma Arterial embolization Hemolytic uremic syndrome Source: Bellomo et al. (2012); KDIGO (2012). Once a diagnosis is made, it is important to classify and stage the severity of the AKI according to the definitions identified earlier in this chapter. However, these definitions for diagnostic criteria should not replace clinical judgment. Early recognition and management of AKI are necessary, as there are associated morbidity and mortality concerns despite advances in renal replacement therapies. Under conditions in which AKI is potentially reversible, a focus should be placed upon management strategies specific to the underlying causes. Continued monitoring and follow-up with resolution and/or progression of AKI will be necessary dependent upon the degree of severity. The first step in the diagnostic process is to determine whether the underlying risk factors obtained from the history and physical examination provide a logical, temporal relationship with AKI. Although it may be useful to classify the severity of AKI through trended changes in urine output and SCr, it is also important to consider the utility and appropriateness of the available diagnostic tests and laboratory assays for assessing the differential of tissue injury specific to the renal anatomy and surrounding areas. Over the past decade, there have been advances in the development of serum and urinary biomarkers targeting renal function and injury. However, the information provided concerning diagnostic testing will focus on more traditional approaches, as further research is necessary before these novel tests can be incorporated into clinical practice. Furthermore, ADQI Consensus Guidelines note that functional and injury biomarkers are predominantly useful in the setting of AKI of uncertain etiology (McCullough et al., 2013). Medications Associated With Acute Kidney Injury MECHANISM DRUG Allergic interstitial nephritis Penicillins, NSAIDs, cephalosporins, sulfonamides, rifampin, allopurinol, cimetidine, phenytoin, vancomycin, ciprofloxacin Decreased renal perfusion Angiotensin-converting enzyme inhibitors, NSAIDs, radiocontrast agents, cyclosporine Direct tubular toxicity Aminoglycosides, amphotericin B, cisplatin, colistin, cyclosporine, foscarnet, pentamidine, heavy metals Rhabdomyolysis Cocaine, lovastatin* Tubular obstruction Acyclovir, sulfonamides, chemotherapeutic agents† Hemolytic uremic syndrome Cyclosporine, cocaine, quinine NSAIDs, nonsteroidal anti-inflammatory drugs. *Rhadomyolysis has been reported with lovastation when combined with cyclosporine. †Uric acid crystals form as a result of tumor lysis. Diagnostic tests and parameters that may help with identifying the potential underlying cause of AKI are listed in Tables 26.4 to 26.6. Table 26.6 presents the parameters for differentiating acute intrinsic kidney injury. Several standard laboratory tests are routinely obtained to properly assess AKI: serum biochemistries, complete blood count (CBC), urinalysis with microscopy, and urine electrolytes. SCr is one of the fundamental laboratory assays utilized as a surrogate of GFR, but is unfortunately an insensitive marker in AKI. This is particularly the case in patients who have a normal baseline renal function and large nephron reserve prior to insult and can readily compensate GFR within homeostasis (Waring & Moonie, 2011). Thus, it is important to consider a patient’s baseline function in comparison to the magnitude of the renal injury. The rise in creatinine may lag behind and may not be detected until sometime after the renal insult. The increase in SCr is typically dependent upon the creatinine tubular secretion, rate of creatinine turnover, and rate of creatinine formation, as well as volume of distribution (Edwards, 2010). The ratio between BUN and SCr can be used to identify type of kidney dysfunction, but is also nonspecific. The BUN/SCr ratio is usually 10:1, but changes in the ratio vary depending on the etiology and severity of AKI. Hyperphosphotemia and hyperkalemia are common signs of AKI, but in the presence of hypercalcemia and hyperuricemia, this may indicate an underlying malignant cause, such as tumor lysis syndrome. Eosinophilia may point to an allergic interstitial nephritis. Tests for immunologic substances such as serum complement 3 and 4 would suggest glomerulonephritis and alert the provider to an autoimmune or systemic cause, such as lupus nephritis.
CHAPTER 26
Acute Kidney Injury
 DEFINITIONS
DEFINITIONS
 EPIDEMIOLOGY
EPIDEMIOLOGY
 ANATOMY, PHYSIOLOGY, AND PATHOLOGY
ANATOMY, PHYSIOLOGY, AND PATHOLOGY
 HISTORY AND PHYSICAL EXAMINATION
HISTORY AND PHYSICAL EXAMINATION
 DIAGNOSTIC CRITERIA
DIAGNOSTIC CRITERIA
Anesthesia Key
Fastest Anesthesia & Intensive Care & Emergency Medicine Insight Engine