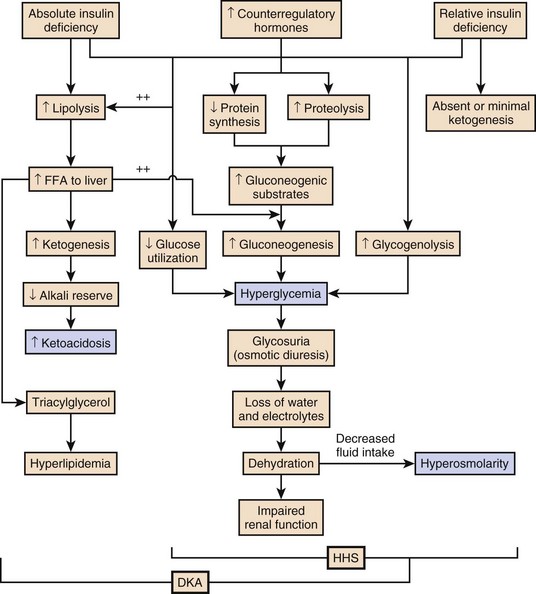
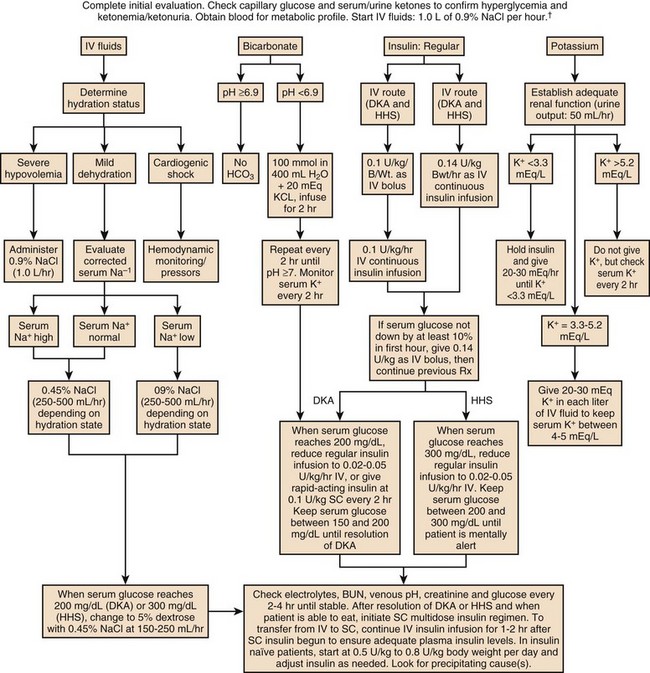
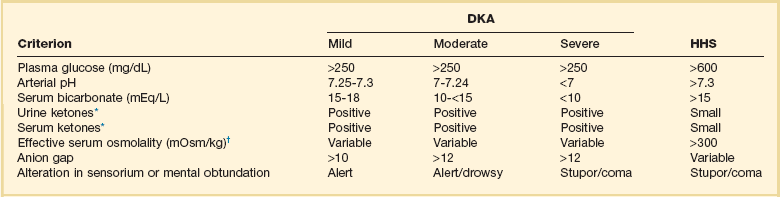
58 DIABETES MELLITUS: EPIDEMIOLOGY AND CLASSIFICATION DIABETIC KETOACIDOSIS AND HYPERGLYCEMIC HYPEROSMOLAR STATE GLYCEMIC CONTROL IN THE INTENSIVE CARE UNIT Epidemiology and Significance of Hyperglycemia Synopsis of Trials of Glycemic Control Technical Issues in Measuring Glucose Current Guidelines for Glycemic Control The Effect of Parenteral Nutrition, Enteral Nutrition, and Glucocorticoids on Glycemic Control Diabetes mellitus (DM) is the fourth to fifth most common cause of death in developed countries and is one of the most common noncommunicable diseases globally.1 DM affects nearly 27 million individuals in the United States and 366 million adults aged 20 to 79 years worldwide.2 Nearly half of those with diabetes are undiagnosed.2 The worldwide incidence of DM is expected to reach 552 million by 2030 with the prevalence of diabetes increasing in every country.2 Diabetes is the leading cause of kidney failure, nontraumatic lower limb amputations, and new cases of blindness among adults in the United States, and is a major cause of heart disease and stroke.3 Diabetes is generally classified as follows: 1. Type 1 diabetes—autoimmune β-cell destruction resulting in an absolute insulin deficiency 2. Type 2 diabetes—progressive defects resulting in increased insulin resistance and a relative insulin deficiency 3. Gestational diabetes—diabetes appearing during pregnancy 4. Diabetes due to other causes—such as genetic defects in β-cell function, genetic defects in insulin action, diseases of the exocrine pancreas (such as cystic fibrosis, pancreatitis, or pancreatectomy), and drug- or chemical-induced diabetes4 Type 2 diabetes accounts for 90% to 95% of DM in the United States.3 The epidemic of type 2 DM has been attributed to the increasing rates of obesity. In patients with diabetes, absolute or relative insulin deficiency leads to hyperglycemia from decreased glucose utilization in peripheral tissues (especially skeletal muscle) and increased hepatic glucose output from glycogenolysis and gluconeogenesis.5 Proteolysis and lipolysis are also increased, providing the amino acids and free fatty acids that are the substrate for gluconeogenesis and alternative fuel sources such as ketones. The various actions of insulin are shown in Box 58.1.6 Diabetic ketoacidosis (DKA) and hyperglycemic hyperosmolar state (HHS) are both part of an overlapping process of decompensated hyperglycemia.7 A significant imbalance between insulin and its counterregulatory hormones (especially glucagon) leads to DKA, a severe metabolic state of hyperglycemia and acidemia from abnormal metabolism of fats, lipids, and carbohydrates.8 DKA is common complication of type 1 DM and is also seen, albeit less frequently, in type 2 DM. The combined findings of serum glucose greater than 250 mg/dL, a pH less than 7.30 with an elevated anion gap, and elevated serum ketones are consistent with DKA. HHS is a state of hyperglycemia associated with osmotic diuresis and dehydration. HHS is defined by glucose levels greater than 600 mg/dL, an elevated serum osmolality, a lack of significant acidemia or ketosis, and typically a change in mental status. HHS has replaced the term “hyperglycemic, hyperosmolar nonketotic coma” because patients may present with a mild elevation in urine or serum ketones and may have a variety of mental status changes other than coma. A careful evaluation of history, physical examination, and laboratory values helps differentiate DKA from HHS (Table 58.1) and assists in distinguishing DKA and HHS from other conditions with overlapping clinical presentation.9 Table 58.1 Diagnostic Criteria for Diabetic Ketoacidosis (DKA) and Hyperglycemic Hyperosmolar Syndrome (HHS) *Nitroprusside reaction method. †Calculation: 2 [measured Na+] + [glucose]/18. Copyright ©2004 American Diabetes Association. Modified from Kitabchi AE, Umpierrez GE, Murphy MB, et al: Hyperglycemic crises in diabetes. Diabetes Care 2004:27:S94-S102, used with permission from the American Diabetes Association. DKA has high morbidity rates, mortality rates, and financial costs despite numerous advances in the treatment of DM. The incidence of DKA in the United States is estimated at 5.9 to 12.9 cases per 100,000 people.10,11 The 120,000 patients admitted to the hospital for DKA each year12 account for approximately $1.4 billion in hospital reimbursement.13 DKA carries a mortality rate of 1% to 5%.9,14 Though the highest mortality rate is in the elderly and in patients with comorbid conditions, DKA remains the leading cause of death in diabetic patients below the age of 24.15 Although most patients will have a history of diabetes, 27% to 37% of patients with DKA carry no prior diagnosis.16,17 This is especially true in young children.18 Although the development of DKA is classically associated with type 1 DM, some patients lack the classic clinical presentation, autoimmune markers, or diminished β-cell reserve of type 1 diabetes.19–21 This subgroup, known as “ketosis-prone type 2 diabetes,” represents patients with type 2 DM who present with DKA and often are African American or Latino, male, middle-aged, and overweight or obese; have a family history of diabetes; and have newly diagnosed diabetes.22,23 Patients with ketosis-prone type 2 DM account for 1 in 5 cases of DKA.22 Patients with “classic” type 2 diabetes may also develop DKA in situations of extreme stress. HHS occurs far less commonly than DKA, with an estimated prevalence of 1 in 1000,24 though the increasing incidence of type 2 DM is expected to increase the prevalence of HHS. HHS typically occurs in patients with type 2 DM and 30% to 40% cases of HHS may be the initial finding leading to diagnosis of type 2 DM.25,26 The mortality rate in HHS tends to be higher than in DKA, with the patient’s comorbid conditions and severity of presentation significantly affecting risk of death.27 The pathophysiology of DKA is driven by insulin deficiency and a rise in the counterregulatory hormones, particularly glucagon, but also epinephrine, cortisol, and growth hormone (Fig. 58.1).28,29 Insulin deficiency results in decreased glucose uptake by peripheral tissues, resulting in elevated serum glucose levels, and hyperglycemia worsens when an increase in glucagon (and an imbalanced insulin/glucagon ratio) results in unrestrained hepatic glycogenolysis and gluconeogenesis.9,24,28 Osmotic diuresis results in total body loss of water and electrolytes, causing intravascular volume depletion. Cellular dehydration occurs as water and electrolytes move from the intracellular to extracellular space. With volume depletion, impaired renal function limits the clearance of glucose and worsens the hyperglycemia.9,24 In an insulin-deficient state proteolysis and lipolysis drive accelerated gluconeogenesis as amino acids and free fatty acids are shunted to the liver.30 At the same time, oxidation of free fatty acids results in the production of the keto acids acetoacetate and β-hydroxybutyrate.9,24 These weak acids react with bicarbonate and diminish the body’s bicarbonate stores. The development of metabolic acidosis in DKA results when the increase in acid production exceeds the body’s buffering capacity.31 HHS is also caused by a relative insulin deficiency and a rise in counterregulatory hormones. Decreased peripheral glucose uptake and increased glycogenolysis and gluconeogenesis result in hyperglycemia followed by osmotic diuresis with volume depletion and dehydration. However, the amount of insulin is sufficient to limit the oxidation of free fatty acids,9 and significant keto acid production does not occur. In the absence of ketoacidosis, the process of dehydration is allowed to continue for longer than in DKA, and patients present with significant hyperosmolarity. The precipitating factors for the development of DKA and HHS are listed in the Table 58.2. Omission of antidiabetic medications is an important factor in the development of DKA and HHS. Omission of insulin may result in DKA in a patient with type 1 diabetes in a matter of hours. In contrast, the development of HHS (or DKA) in a patient with type 2 diabetes occurs over days to weeks in a patient omitting insulin or oral agents. In addition to the factors listed in the table, impaired thirst or lack of access to water, particularly in the elderly, will contribute to the development of HHS.32 Eating disorders may account for up to 20% of cases of recurrent DKA.9 Table 58.2 Causes of Diabetic Ketoacidosis and Hyperglycemic Hyperosmolar Syndrome Superscript numbers refer to references provided online for this chapter. The presentation of DKA and HHS varies with the severity of the metabolic derangement and any intercurrent illnesses or comorbid conditions. DKA can occur in a patient without a history of diabetes who presents with only mild symptoms; a high index of suspicion is needed to make the diagnosis in this setting, particularly in children under the age of 6. A detailed evaluation should reveal precipitating factors, especially infection and medication nonadherence, which are common triggers of DKA and HHS (see Table 58.2).24 Patients may present with symptoms of hyperglycemia (polyuria, polyphagia, polydipsia, or blurred vision) or with nonspecific symptoms of fatigue, abdominal pain, nausea, vomiting, and headache. Mental status can vary from somnolence to lethargy and even coma as the acidosis or hyperosmolarity worsens. Volume depletion may be manifested by tachycardia, poor skin turgor, dry mucous membranes, and orthostatic hypotension. In patients with DKA, metabolic acidosis causes rapid deep breathing (known as Kussmaul respiration), and acetone (derived from acetoacetate) can be sensed as a fruity smell on the patient’s breath. The diagnosis of DKA is based on a serum glucose level greater than 250 mg/dL, presence of ketones, arterial pH less than 7.3, and a CO2 concentration less than 18 mmol/L.5,9 The anion gap will be elevated, reflecting the presence of the unmeasured keto acids. The laboratory findings and changes in mental status help the clinician gauge the severity of DKA and distinguish DKA from HHS. Although arterial blood had previously been the standard for determining pH, current evidence supports the use of venous pH as a less expensive, more accessible alternative.33,34 Exceptions to the typical presentation include pregnant patients or those with poor oral intake who may have a serum glucose value below 250 mg/dL, or even in the normal range.35,36 In patients with severe vomiting, the bicarbonate value may be normal or elevated. The serum potassium level may also be normal, low, or elevated depending on factors such as the degree of acidosis or osmotic diuresis and the duration of the process. Insulin deficiency and acidosis decrease cellular uptake of potassium, although hyperosmolality enhances cellular efflux of potassium.37 Large amounts of potassium shift from the intracellular space to the extracellular space and are then lost in the urine. Regardless of the serum potassium level, all patients with DKA and HHS have substantial depletion of total body potassium.9,37,38 The serum osmolality (in mOsm/kg) can be estimated by the following calculation: Serum osmolarity is generally normal or mildly elevated in DKA but is often profoundly elevated in patients with HHS. Semiquantitative nitroprusside assays for urine and serum ketones are commonly employed but test only for acetone and acetoacetate, not β-hydroxybutyrate. This can be problematic, although β-hydroxybutyrate is the predominant keto acid, accounting for 75% of the keto acid load in DKA.39 In addition, nitroprusside assays may remain positive long after resolution of the metabolic acidosis, so serial measurements may be misleading and are not recommended. Quantitative assays for β-hydroxybutyrate are available and may be useful for diagnosis, especially if the nitroprusside test is negative. A small pilot study found that a serum level above 3.5 mmol/L has 100% specificity for DKA.40 Unlike the nitroprusside assay, the β-hydroxybutyrate test can also be used to monitor the management of DKA because appropriate treatment will be accompanied by a progressive fall in β-hydroxybutyrate levels. The diagnostic criteria that distinguish HHS from DKA include higher plasma glucose (>600 mg/dL), a lack of acidosis (pH > 7.3, CO2 > 18 mEq/L), more profound dehydration (serum osmolality > 320 mOsm/kg), and a change in mental status.9 Patients with HHS often have low levels of ketones (β-hydroxybutyrate between 0.3 and 3.0 mmol/L) compared with DKA patients, whose β-hydroxybutyrate levels will be greater than 3.0 mmol/L. Initial laboratory studies should include electrolytes, phosphorus, blood urea nitrogen (BUN), creatinine, urinalysis, complete blood count with differential, and electrocardiogram (see Table 58.2). Further evaluation should be undertaken based on the patient’s presentation. A variety of laboratory abnormalities are frequently seen in DKA. Amylase, from both salivary and pancreatic sources, may be increased in up to 90% of patients with DKA. Lipase, normally a sensitive marker for pancreatitis, may also be elevated in DKA. However, the clinician should be aware that 10% to 15% of DKA patients do have concomitant pancreatitis.41,42 Leukocytosis of 10,000 to 15,000 white blood cells/mm3 is common, but levels greater than 25,000 cells/mm3 or the presence of greater than 10% band neutrophils should increase the clinical suspicion for an active infection. Elevated hemoglobin due to the volume depletion may be present. High liver function studies occur commonly, especially in patients with fatty liver,43 and mild increases in creatine kinase and troponin may occur in the absence of myocardial damage.44 Intercurrent illnesses contribute to the morbidity and mortality rates in a hyperglycemic emergency, and every patient with DKA or HHS should undergo a thorough assessment with a focus on infectious and cardiovascular causes.34 Appropriate management should be started immediately and continue concurrent to the treatment of the DKA and HHS. Additional causes of acidosis or ketosis should be considered in the differential diagnosis including starvation ketosis; alcoholic ketoacidosis; lactic acidosis; intoxication from methanol, salicylate, or ethylene glycol; chronic renal failure; and rhabdomyolysis.34 The treatment of HHS and DKA in adults and children follows similar principles of intravenous (IV) fluid, insulin, and electrolyte management. Children require more attention to weight-based regimens for fluid management in order to avoid cerebral edema.45 The details of the management of HHS and DKA in patients older than 20 years are detailed in Figure 58.2. Although hyperglycemia drives the cascade of osmotic diuresis, volume depletion, and renal insufficiency, the process is reversed with appropriate fluid replacement.46 In both DKA and HHS, normal saline solution should be started at 15 to 20 mL/kg body weight per hour initially (maximum of 1 L/hour), with hourly assessments of fluid status and urine output.46 The infusion rate can be lowered to 250 to 500 mL/hour once the blood pressure stabilizes. An initial bolus of regular insulin is typically given at 0.1 to 0.15 units/kg body weight followed by an IV insulin infusion at 0.1 unit/hour, though there is little evidence to support the need for an initial bolus.47,48 Higher dose insulin regimens are not beneficial.49,50 In patients receiving IV insulin, point of care testing should be performed every 1 to 2 hours and may be changed to every 2 hours once the patient is stable. Although subcutaneous (SC) and intramuscular (IM) insulin protocols can treat DKA or HHS, the initial response to glucose lowering is slower, and SC or IM regimens are generally reserved for emergent situations (i.e., a DKA patient with severe hyperkalemia and electrocardiographic [ECG] changes) in which IV access has been delayed.49,51 With the initial correction of volume depletion, particularly in HHS, the glucose level may fall precipitously. Thereafter, glucose levels should make a steady decline of 50 to 75 mg/dL each hour.34 There is no advantage to a more rapid improvement in the glucose level. The rate of insulin infusion may be adjusted to allow the glucose to trend downward at an appropriate rate. Once the glucose level decreases to less than 250 mg/dL, dextrose is added to the IV fluid to allow for the continued insulin infusion necessary to resolve the acidosis, because the fall in glucose will precede normalization of the acid-base status.9 By this point, the insulin infusion rate will usually have been adjusted to 0.02 to 0.05 unit/hour. The insulin infusion should be continued until resolution of the hyperglycemic crisis. In DKA, resolution of ketoacidosis is indicated by a normalization of the anion gap or a drop in the β-hydroxybutyrate level to less than 3 mmol/L.52 In HHS, resolution is indicated by correction of the fluid and electrolyte abnormalities. Once the acute abnormalities have resolved and the glucose level has improved to less than 200 mg/dL, the patient can be transitioned to scheduled SC insulin. Patients should be placed on a physiologic insulin regimen, which includes a basal insulin (NPH, glargine, or detemir) and a mealtime, or “nutritional,” insulin (regular, lispro, aspart, or glulisine). NPH and regular insulin are associated with higher rates of hypoglycemia.53 The use of corrective (“sliding scale”) insulin as the sole SC insulin strategy is never appropriate in a patient recovering from DKA. Most protocols call for 80% to 100% of the calculated daily insulin requirements to be started based on a stable insulin infusion.54–57 The insulin doses are divided into 50% basal and 50% nutritional insulin. Older age, higher IV insulin requirements, and greater blood glucose variation decrease the chance of a successful transition.56 Other potential pitfalls include discontinuing the IV insulin without initiating physiologic insulin, and over- or underestimating the 24-hour insulin requirements based on fluctuating insulin infusion rates. Despite pressures in a busy intensive care unit (ICU) to transition patients off IV insulin, the SC insulin needs to be calculated based on the relatively stable IV insulin requirements over 4 to 6 hours prior to the transition.57 IV insulin should not be discontinued until at least 2 hours after the start of basal insulin. A protocol for the successful transition from IV to SC insulin is listed in Box 58.2.58 Box 58.2 Important Steps in Transition from Insulin Infusion to Subcutaneous (SC) Insulin Step 1: Is the patient stable enough for transition? Step 2: Does the patient need to switch to scheduled SC insulin? Step 3: If transition is needed, calculate a total daily dose (TDD) of insulin. The TDD is an estimate of the 24-hour insulin requirement when the patient is receiving full nutrition. • Determine mean insulin infusion rate from last 6-8 hours. • Calculate 24-hour insulin dose based on this rate, and reduce the 24-hour dose by 20%. • Multiply hourly insulin infusion rate by 20 (80% of 24) to arrive at a safety-adjusted 24-hour insulin dose. • Determine if this total is the TDD or basal dose based on current nutrition. Step 4: Construct a basal/bolus regimen tailored to the patient’s nutritional situation, building safeguards for any changes in nutritional intake and uncertainties about reliability of intake. Several options are again available. • Basal: glargine, detemir, or NPH • Nutritional: The remainder of TDD is scheduled nutritional insulin in divided doses. In general, these doses need to be adjusted down for <100% nutritional intake, and the orders should allow for administering nutritional insulin just AFTER observed meal to allow an assessment of intake. Several options are available for estimating the initial doses: Step 5: Be sure to give SC insulin BEFORE the infusion stops. DM, diabetes mellitus; HbA1c, hemoglobin A1c (glycosylated hemoglobin). Modified from O’Malley CW, Emanuele, M, Halasyamani L, Amin AN. Bridge over troubled waters: safe and effective transitions of the inpatient with hyperglycemia. J Hops Med 2008, 3(S5):S55-65. Patients with DKA or HHS may initially present with hyperkalemia or hypokalemia, each with significant potential cardiac consequences. Tall peaked T waves in the precordial leads are the first ECG sign of hyperkalemia as the potassium increases above 5.5 mmol/L. As potassium levels increase to greater than 6.5 mmol/L, additional ECG changes emerge: prolonged PR interval, decreased amplitude of P waves, and a widening of the QRS complex. As the potassium increases to greater than 8.0 mmol/L more ominous changes occur, including loss of P waves; intraventricular, fascicular, and bundle branch blocks; and widening of the QRS complex, ultimately resulting in asystole.59 Conversely, hypokalemia also increases the risk of cardiac arrhythmia. ECG changes in hypokalemia include broad flat T waves, ST-segment depression, and QT interval prolongation.60 Additionally, hypokalemia decreases myocardial contractility.60 Treatment with volume expansion and insulin will lower serum potassium levels as potassium shifts from the extracellular to intracellular space. Electrolytes should be monitored every 2 to 4 hours in the early stages of DKA.9,32 For a potassium concentration greater than 5 mmol/L, serum K+ testing should be repeated every 1 to 2 hours. Potassium replacement should be started once the potassium decreases into the normal range and the patient is producing urine. Once serum potassium is below 5 mmol/L, patients should receive KCl 5 mEq/hour via the IV fluid, and the rate can be increased to 10 mEq/hour if the serum K+ is below 4 mmol/L. Lower doses should be used if the patient has evidence of impaired renal function. The metabolic acidosis of DKA impairs myocardial contractility, affects oxyhemoglobin dissociation and tissue oxygen delivery, inhibits intracellular enzymes, alters cellular metabolism, and may result in vital organ dysfunction.61 Correction of acidosis focuses on the correction of insulin deficiency and volume depletion, as well as any other underlying conditions that may be contributing to the acidosis. Although the administration of bicarbonate would appear to be a reasonable consideration, multiple trials of bicarbonate replacement in DKA patients have demonstrated no benefit.61,62 Furthermore, the use of IV bicarbonate may result in complications.34,40 Bicarbonate therapy has been associated with hypokalemia, intracellular acidosis, and tissue hypoxia.47 On the other hand, a potential benefit for bicarbonate therapy in those individuals with severe acidemia (pH < 6.9)62,63 has been suggested, though the literature is limited. In patients with pH less than 6.9, the use of sodium bicarbonate (100 mmol sodium bicarbonate in 400 mL sterile water with 20 mEq KCl, administered at a rate of 200 mL/hour for 2 hours, and then repeating every 2 hours) may be considered until the pH is greater than 6.9.9 Phosphate levels initially may be normal to elevated, but usually decline with treatment.64 There is no evidence to support the routine use of phosphate replacement in patients with DKA.65,66 Nevertheless, severe hypophosphatemia may impair oxygen delivery and cause muscle fatigue, and selective replacement is suggested in patients with phosphate levels less than 1.0 mg/dL, anemia, respiratory failure, or CHF.9,67 Correcting the phosphorus level improves tissue oxygenation and restores the body’s buffering capacity. Phosphate is generally replaced as a potassium salt.68 To avoid complications, particularly hypocalcemia, phosphorus replacement should be limited to 40 to 50 mmol of potassium phosphate at approximately 3 to 4 mmol/hour. The potassium being provided in other IV fluids may need to be adjusted to account for the potassium infused as part of the phosphorus replacement.68 The development of hyperchloremic acidosis occurs in DKA because the ability to completely regenerate bicarbonate is hampered by the urinary loss of keto acids, which normally act as substrate for the re-formation of bicarbonate. To maintain the appropriate balance of cations and anions, chloride ions from the infused saline will partially compensate for the lack of bicarbonate. As the ketoacidosis resolves, a hyperchloremic acidosis (with a normal anion gap) may ensue. This benign process resolves gradually in the hours to days after the saline infusion is reduced or stopped.68 Cerebral edema is the one of the most feared complications of hyperglycemic crisis. Classically, cerebral edema has been reported in children with DKA. It occurs in 0.5% to 1% of all DKA cases69,70 and carries a mortality rate of 20%. Survivors are at risk for residual neurologic problems.45 Although cerebral edema is occasionally seen in adults with HHS, it is rare in adults with DKA and children with HHS.71 Children who present with (1) low partial pressures of arterial carbon dioxide and (2) high serum urea nitrogen concentrations and (3) are treated with bicarbonate are at increased risk for cerebral edema. The exact cause of cerebral edema remains unknown. It does not appear to be associated with the initial level of hyperosmolarity or more rapid correction of the hyperosmolarity.72,73 Cerebral ischemia, and not changes in osmolality, may be the causal factor in patients who develop cerebral edema.73 Nevertheless, care should be taken to avoid rapid changes in glucose and osmolarity, especially in younger patients. The clinician should also be aware of the signs of cerebral edema, which include headache, persistent vomiting, hypertension, bradycardia, and neurologic changes. Attention to the diagnosis and management of any infections or cardiovascular events is imperative. Other complications of the management of DKA and HHS include hypokalemia, hypophosphatemia, acute renal failure, and shock. Appropriate management of hyperglycemic emergencies with close attention to fluid and electrolyte balance minimizes these complications. Less common problems can include rhabdomyolysis,74 thrombosis and stroke,75 pneumomediastinum,76 and memory loss with decreased cognitive function in children.77 Despite improvements in insulin treatment and glucose monitoring78 the incidence of DKA is increasing.3 Prevention is paramount. Health care providers should recognize signs of DM in all age groups. Patients with DM and their caregivers should be familiar with sick day recommendations (Box 58.3), and patients with type 1 DM should check urine ketones by dipstick if the glucose is greater than 240 mg/dL.79 Home measurement of serum ketones with a commercial glucometer may allow earlier detection of DKA and decreased hospital visits,80 though most meters lack this capability. Box 58.3 Sick Day Management Protocol for Diabetic Patients212,213 • Check blood sugar levels at least every 4 hours, but when values are changing quickly, check more often. • Check urine or blood ketones. • Modify usual insulin regimen according to a plan developed by the diabetes physician or team. • Maintain adequate food and fluid intake. If your appetite is poor, aim for consumption of 50 g of carbohydrate every 3-4 hours. If you are nauseous, high-carbohydrate liquids, such as regular (not diet) soft drinks or juice, or frozen juice bars, sherbet, pudding, creamed soups, or fruit-flavored yogurt usually are tolerated. Broth also is a good alternative. Taking Medications When You Are Sick Examples of When to Call Physician or Diabetes Team
Acute Diabetic Emergencies, Glycemic Control, and Hypoglycemia
Diabetes Mellitus: Epidemiology and Classification
Diabetic Ketoacidosis and the Hyperglycemic Hyperosmolar State
Epidemiology
Pathophysiology
Precipitating Factors
Etiologic Factor/Condition
Diagnoses with Comments
Inadequate insulin
Nonadherence,25,193,194 inadequate insulin regimen,34 insulin pump failure or omission of insulin,195,196 new-onset diabetes197
Infections
Any moderate to severe infectious process29,198
Most commonly, pneumonia, urinary tract infection, sepsis9
Acute medical conditions
Myocardial infarction,9 pancreatitis,41 cerebrovascular accident,9 arterial thrombosis (mesenteric, iliac)199
Interference with glucose metabolism or insulin secretion32
Thiazide diuretics, corticosteroids,200 second-generation (atypical) antipsychotics,201 FK506,202 glucagon,203 sympathomimetic agents (albuterol, terbutaline, dobutamine)204
Endocrine disturbance
Pheochromocytoma,205 hyperthyroidism,206 acromegaly,207 hemochromatosis208
Substance abuse
Cocaine209
Nutritional factors
Parenteral nutrition210
Immune modulation
Interferon211
Presentation
Diagnosis
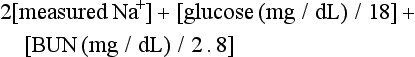
Management
Fluid Replacement
Insulin

Potassium
Bicarbonate
Phosphate
Hyperchloremic Acidosis
Complications of the Management of Hyperglycemic Emergencies
Prevention