76 Acute Coronary Syndromes
Therapy
 Definition and Clinical Manifestations
Definition and Clinical Manifestations Pathophysiology of Acute Coronary Syndromes
Pathophysiology of Acute Coronary Syndromes
The inciting event underlying the development of an ACS is rupture of an atherosclerotic plaque.1 Possible sequelae of plaque rupture include thrombus formation with total occlusion, with likely development of STEMI; dissolution of thrombus and healing of the fissure, with clinical stabilization; and subtotal occlusion, which can lead to either NSTEMI or UA.
Atherosclerotic plaques are composed of a lipid core that includes cholesterol, oxidized low-density lipoproteins (LDL), macrophages, and smooth muscle cells, covered by a fibrous cap. Plaque rupture occurs when external mechanical forces exceed the tensile strength of the fibrous cap. After plaque rupture, the clinical consequences depend largely on the balance between prothrombotic and antithrombotic forces.2 The lipid core contains tissue factor and other thrombogenic materials that lead to platelet activation and aggregation. Fibrinolytic factors such as tissue plasminogen activator, prostacyclin, and nitric oxide act to counteract the potential for thrombosis. A major factor in the outcome of plaque rupture is blood flow. With subtotal occlusion, high-grade stenosis, or vasospasm, thrombus begins to propagate downstream in the arterial lumen. In contrast to the initial thrombi, which are platelet rich, these thrombi contain large numbers of red cells enmeshed in a web of fibrin. The former would be expected to respond best to antiplatelet therapy, the latter to antithrombotic and fibrinolytic therapy.
 ST-Segment Elevation Myocardial Infarction
ST-Segment Elevation Myocardial Infarction
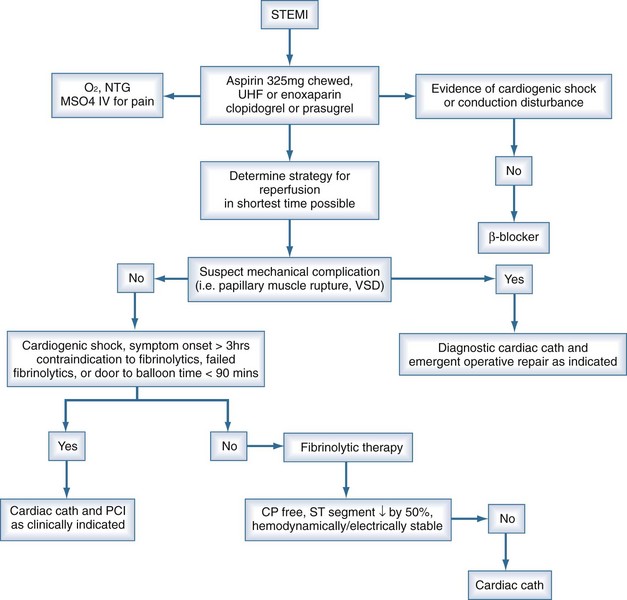
Patients presenting with suspected myocardial ischemia should undergo a rapid evaluation and should be treated with oxygen, sublingual nitroglycerin (unless systolic pressure is <90 mm Hg), and aspirin, 160 to 325 mg orally.3,4 Opiates relieve pain and also reduce anxiety, the salutary effects of which have been known for decades and should not be underestimated. A 12-lead ECG should be performed and interpreted expeditiously. Figure 76-1 shows a possible treatment algorithm for patients with STEMI.
ST-segment elevation of at least 1 mV in two or more contiguous leads provides strong evidence of thrombotic coronary occlusion, and the patient should be considered for immediate reperfusion therapy. The diagnosis of STEMI can be limited in the presence of preexisting left bundle branch block (LBBB) or permanent pacemaker. Nonetheless, new LBBB with a compatible clinical presentation should be treated as acute myocardial infarction (AMI) and treated accordingly. Indeed, recent data suggest that patients with STEMI and new LBBB may stand to gain greater benefit from reperfusion strategies than those with ST elevation.5
Fibrinolytic Therapy
Early reperfusion of an occluded coronary artery is indicated for all eligible candidates. Overwhelming evidence from multiple clinical trials demonstrates the ability of fibrinolytic agents administered early in the course of an acute MI to reduce infarct size, preserve LV function, and reduce short-term and long-term mortality.6–8 Patients treated early derive the most benefit.9 Multiple studies conclude that greatest mortality benefit is seen if fibrinolytics are administered within the first 12 hours of symptom onset,8,10,11 but it is reasonable to administer fibrinolytics to patients whose onset of symptoms exceeds 12 hours but who have continued clinical or ECG evidence of ischemia.
Indications for and contraindications to fibrinolytic therapy are listed in Box 76-1. Because of the small but nonetheless significant risk of a bleeding complication, most notably intracranial hemorrhage, selection of patients with AMI for administration of a fibrinolytic agent should be undertaken with prudence and caution. That is of special importance in ICU patients who may have a predisposition to bleeding complications because of multiple factors. Contraindications can be regarded as absolute or relative. In the surgical patient, thrombolysis may pose a prohibitive risk, and emergent coronary angiography (with percutaneous coronary intervention [PCI] as clinically indicated) may be preferable.
Box 76-1
Indications for and Contraindications to Fibrinolytic Therapy in Acute Myocardial Infarction
Indications
After administration of fibrinolytics for STEMI, the patient should be monitored for signs and symptoms of adequate reperfusion within 90 minutes, as indicated by relief of symptoms and/or hemodynamic/electrical instability coupled with at least a 50% resolution of the highest initial ST elevation.12,13 If signs of adequate reperfusion are not evident within 90 minutes, patients should be taken to the cardiac catheterization lab and considered for PCI. More recent data support the notion that all patients who receive fibrinolytics for STEMI and have at least one high-risk feature should have cardiac catheterization for risk stratification and potential percutaneous revascularization,14,15 even if this involves immediate transfer from the presenting hospital to a PCI-capable facility. Patients not considered high-risk may be observed in the initial facility where fibrinolytics were administered. High-risk features include extensive ST-segment elevation (>2 mm ST elevation in two anterior leads), new-onset LBBB, previous MI, Killip class 2 or 3 or left ventricular ejection fraction (LVEF) ≤ 35%, systolic blood pressure ≤ 100 mm Hg, heart rate ≥ 100 bpm, or right ventricular involvement.
In contrast to the treatment of STEMI, fibrinolytics have shown no benefit and an increased risk of adverse events when used for the treatment of UA/NSTEMI.16 Based on these findings, there is currently no role for fibrinolytic agents in these latter syndromes.
Fibrinolytic Agents
Streptokinase was the original lytic agent used in MI, but it has now been superseded by tissue plasminogen activator (tPA),6 which is more fibrin selective than streptokinase and produces a higher early coronary patency rate (70%-80%).17,18 Administration of tPA usually follows an accelerated regimen consisting of a 15-mg bolus, 0.75 mg/kg (up to 50 mg) IV over the initial 30 minutes, and 0.5 mg/kg (up to 35 mg) over the next 60 minutes. Reteplase (rPA) is a deletion mutant of tPA with an extended half-life, and is given as two 10-U boluses 30 minutes apart. Reteplase was originally evaluated in angiographic trials that demonstrated improved coronary flow at 90 minutes compared to tPA, but subsequent trials showed similar 30-day mortality rates.19 Tenecteplase (TNK-tPA) is a genetically engineered tPA mutant with amino acid substitutions that result in prolonged half-life, resistance to plasminogen activator inhibitor 1, and increased fibrin specificity. TNK-tPA is given as a single bolus adjusted for weight. A single bolus of TNK-tPA has been shown to produced coronary flow rates identical to those seen with accelerated tPA, with equivalent 30-day mortality and bleeding rates.20
Primary Percutaneous Coronary Intervention in Acute Myocardial Infarction
The major advantages of primary PCI over fibrinolytic therapy include a higher rate of normal flow (TIMI grade 3),7 lower risk of intracranial hemorrhage, and the ability to stratify risk based on the severity and distribution of coronary artery disease. Patients ineligible for fibrinolytic therapy should obviously be considered for primary PCI. In addition, data from several randomized trials have suggested that PCI is preferable to fibrinolytic therapy for several subsets of AMI patients at higher risk.21,22 The largest of these trials is the GUSTO-IIb Angioplasty Substudy, which randomized 1138 patients. At 30 days, there was a clinical benefit in the combined primary endpoint of death, nonfatal reinfarction, and nonfatal disabling stroke in the patients treated with percutaneous transluminal coronary angioplasty (PTCA) compared to tPA, but no difference in the “hard” endpoints of death and MI at 30 days.22
Recent meta-analyses comparing direct PTCA with fibrinolytic therapy have suggested lower rates of mortality and reinfarction among those receiving direct PTCA.23,24 Thus direct angioplasty, if performed in a timely manner (ideally within 60 minutes) by highly experienced personnel, may be the preferred method of revascularization, since it offers more complete revascularization with improved restoration of normal coronary blood flow and detailed information about coronary anatomy.3 There are certain subpopulations in which primary PCI is clearly preferred, and other populations in which the data are suggestive of benefit. These subsets are listed in Box 76-2. More important than the method of revascularization is the time to revascularization, and that this should be achieved in the most efficient and expeditious manner possible.25 It is important to keep in mind that early, complete, and sustained reperfusion after MI is known to decrease 30-day mortality. The preferred method for reperfusion in STEMI is PCI only if it can be done within a timely manner. Practical considerations regarding transport to a PCI-capable facility should be carefully reviewed before forgoing thrombolytics for PCI. Early recognition and diagnosis of STEMI are key to achieving the desired door-to-needle (or medical contact–to-needle) time for initiation of fibrinolytic therapy of 30 minutes or door-to-balloon (or medical contact–to-balloon) time for PCI under 90 minutes.3 Achieving reperfusion in timely matter correlates with improvement in ultimate infarct size, LV function, and survival.12,13 The ultimate goal is to restore adequate blood flow through the infarct-related artery to the infarct zone, as well as to limit microvascular damage and reperfusion injury. The latter is accomplished with adjunctive and ancillary treatments that will be discussed in the following sections.
Box 76-2
Situations in Which Primary Angioplasty is Preferred in Acute Myocardial Infarction
Coronary Stenting
Primary angioplasty for AMI results in a significant reduction in mortality but is limited by the possibility of abrupt vessel closure, recurrent in-hospital ischemia, reocclusion of the infarct related artery, and restenosis. The use of coronary stents has been shown to reduce restenosis and adverse cardiac outcomes in both routine and high-risk PCI.26 The PAMI Stent Trial was designed to test the hypothesis that routine implantation of an intracoronary stent in the setting of MI would reduce angiographic restenosis and improve clinical outcomes compared to primary balloon angioplasty alone. This large, randomized, multicenter trial involving 900 patients did not show a difference in mortality at 6 months but did show improvement in ischemia-driven target vessel revascularization and less angina in the stented patients compared to balloon angioplasty alone.27 Despite the lack of definite data demonstrating mortality benefit, virtually all the trials investigating adjunctive therapy for STEMI have employed a strategy of primary stenting, and stenting has becoming the default strategy. Whether to use a bare metal stent or a drug-eluting stent in acute MI is a question that has not yet been addressed definitively by clinical trials; selection is currently based on both patient and angiographic characteristics.
Adjunctive Therapy to Primary PCI
Aspirin
Aspirin is the best known and the most widely used of all the antiplatelet agents because of low cost and relatively low toxicity. Aspirin inhibits the production of thromboxane A2 by irreversibly acetylating the serine residue of the enzyme prostaglandin H2 synthetase. Aspirin has been shown to reduce mortality in acute infarction to the same degree as fibrinolytic therapy, and its effects are additive to fibrinolytics.28 In addition, aspirin reduces the risk of reinfarction.29,30 Unless contraindicated, all patients with a suspected ACS (STEMI, NSTEMI, UA) should be given aspirin as soon as possible.
Thienopyridines
Thienopyridines are a class of oral antiplatelet agents that block the P2Y12 component of the adenosine diphosphate receptor and thus inhibit the activation and aggregation of platelets. Currently used thienopyridines include clopidogrel and prasugrel.32 Clopidogrel is a prodrug that is converted in the liver to the active thiol metabolite via the cytochrome P450 (CYP) 3A, 1A, 2B, and 2C subfamilies. The active metabolite irreversibly binds to the P2Y12 component of the ADP receptor on the platelet surface, which prevents activation of the GPIIb/IIIa receptor complex and reduces platelet aggregation for the remainder of the platelet’s lifespan, approximately 7 to 10 days. Onset of inhibition of platelet aggregation (IPA) is dose dependent, with a 300- to 600-mg loading dose achieving inhibition of platelet within 2 hours, whereas a dose of 50 to 100 mg achieves inhibition of platelets in about 24 to 48 hours. Peak effect (time to maximal IPA) occurs at 6 hours with a loading dose of 300 to 600 mg31 and 5 to 7 days with a dose of 50 to 100 mg.32
The efficacy of clopidogrel in combination with aspirin administered to patients with STEMI prior to PCI was tested in the COMMIT-CCS 2 and CLARITY TIMI-28 studies. CLARITY TIMI-2833 randomized 3491 STEMI patients to clopidogrel (300-mg load followed by 75 mg daily) or placebo. All patients also received a fibrinolytic, aspirin, and when appropriate, heparin. Use of clopidogrel decreased the incidence of the primary composite efficacy endpoint (infarct artery patency or death or recurrent MI before angiography, 15.0 % versus 21.7%, P < 0.001), largely due to a difference in occlusion of the infarct-related artery (12% versus 18%), with no difference in mortality or major bleeding. In the 1863 patients in CLARITY TIMI-28 who underwent PCI (reported as CLARITY-PCI), retreatment with clopidogrel prior to PCI for STEMI resulted in a significant reduction in cardiovascular death, MI, or stroke at 30 days (7.5% versus 12.0%; P = 0.001) without causing excess bleeding.34 It is therefore routine practice to administer a loading dose of clopidogrel, 300 mg or 600 mg, prior to PCI regardless of the physician’s concern that the patient might need coronary artery bypass graft (CABG) in the near future.
Some patients are considered clopidogrel nonresponders, usually defined as a recurrence of cardiovascular events while on the recommended dose. Ex vivo assays measuring the degree of inhibition of platelet aggregation while on clopidogrel have demonstrated that 4% to 30% of patients do not have an adequate platelet response while on clopidogrel.37–39 Despite these findings, testing for clopidogrel resistance has not become routine.
Prasugrel is a recently approved thienopyridine that irreversibly binds to the P2Y12 component of the ADP receptor with a more rapid onset of action.40 Like clopidogrel, prasugrel is a prodrug metabolized to both an active and inactive metabolite, but a higher proportion is metabolized to an active metabolite, resulting in a higher level of inhibition of platelet aggregation than clopidogrel. The onset of inhibition of platelet aggregation is dose dependent and can be achieved in less than 30 minutes at a dose of 60 mg, but peak effect of IPA occurs in approximately 4 hours.35 The randomized double-blind TRITON-TIMI 38 trial compared prasugrel (loading dose of 60 mg followed by maintenance dose of 10 mg) with clopidogrel (300-mg load followed by 75-mg maintenance) in 13,608 patients with UA/NSTEMI (n = 10,074) or STEMI (n = 3534) who underwent PCI.36 All patients also received aspirin, and treatment with prasugrel or clopidogrel was continued for a median of 14.5 months. The primary endpoint, a composite of cardiovascular death, nonfatal MI, and nonfatal stroke, was less frequent among patients who received prasugrel (9.9% versus 12.1 %, P < 0.001). The rate of major bleeding was higher in the prasugrel group (2.4% versus 1.8 %, P = 0.03), as was the rate of life-threatening bleeding. A post hoc analysis of the TRITON TIMI-38 trial identified three ACS subgroups in which prasugrel was found to be harmful or showed no net benefit: patients with a history of transient ischemic attack (TIA) or stroke (net harm), age older than 75 (no net benefit), and body weight less than 60 kg ( no net benefit). The FDA has labeled history of TIA and/or stroke as a contraindication to prasugrel use.36
Dual antiplatelet therapy with aspirin and thienopyridines is given to all patients undergoing PCI, as described above. However, data suggest that even patients not undergoing PCI benefit from the addition of clopidogrel to aspirin. COMMIT-CCS-2 randomized over 45,000 patients with suspected MI to 75 mg of clopidogrel daily (no loading dose).37 The majority of patients had STEMI, but only 54% were treated with fibrinolytics. Clopidogrel was continued after hospital discharge for a mean duration of 14.9 days. The co-primary endpoint of all-cause mortality was reduced from 8.1% in the placebo group to 7.5% in the clopidogrel group (OR, 0.93 [95% CI, 0.87-0.99]; P = 0.03; NNT = 167), without increased bleeding in the clopidogrel group. On the basis of these data, patients presenting with MI should be considered for a thienopyridine regardless of whether or not they underwent reperfusion therapy. The duration of thienopyridine use in this population has yet to be defined.
Glycoprotein IIb/IIIa Receptor Antagonists
Glycoprotein IIb/IIIa receptor antagonists inhibit the final common pathway of platelet aggregation, blocking cross-linking of activated platelets, and are often-used percutaneous interventions.38–42 In the era of dual antiplatelet therapy using a thienopyridine and aspirin, the role of addition of a glycoprotein IIb/IIIa inhibitor in primary angioplasty for STEMI is uncertain. Studies such as the ADMIRAL and CADILLAC trials conducted prior to the use of dual antiplatelet therapy established the efficacy of abciximab in primary PCI (with or without stenting) in patients with STEMI.41 The results of recent clinical trials have raised questions about whether glycoprotein IIb/IIIa antagonists have additional utility when added to dual antiplatelet therapy in patients with STEMI.43–45 The BRAVE-3 trial randomized 800 patients undergoing primary stenting to 600 mg of clopidogrel plus either placebo or abciximab prior to PCI and showed no difference at 30 days in either the primary endpoint of infarct size or the secondary composite endpoint of death, recurrent MI, stroke, or urgent revascularization of the infarct-related artery.43 Similar findings were seen in ON-TIME2, in which 984 patients with STEMI were randomized to either high-dose tirofiban or placebo in addition to dual antiplatelet therapy prior to transport for PCI. Although patients who received high-dose tirofiban had improved resolution of ST-segment elevation before and after PCI, there was no significant difference in TIMI flow or the 30-day composite endpoint of death, recurrent MI, or urgent target-vessel revascularization between the two groups.44 Given the present data, current guidelines suggest that when a STEMI patient is treated with a thienopyridine and aspirin plus an anticoagulant such as UFH or bivalirudin, the use of a glycoprotein IIb/IIIa inhibitor at the time of PCI may be beneficial but cannot be recommended as routine.3
Anticoagulants
Administration of full-dose heparin after fibrinolytic therapy with tPA is essential to diminish reocclusion after successful reperfusion.6,28 Dosing should be adjusted to weight, with a bolus of 60 U/kg up to a maximum of 4000 U and an initial infusion rate of 12 U/kg/h up to a maximum of 1000 U/h, with adjustment to keep the partial thromboplastin time (PTT) between 50 and 70 seconds.4 Heparin should be continued for 24 to 48 hours. For patients undergoing PCI who have already been treated with aspirin and a thienopyridine, both unfractionated heparin or bivalirudin (with or without prior heparin administration) are acceptable anticoagulant regimens.3 Bivalirudin is a direct thrombin inhibitor that inhibits both clot-bound and circulating thrombin. It is administered as an initial bolus of 0.75 mg/kg, followed by a continuous infusion at 1.75 mg/kg/h for the duration of PCI, with adjustments for patients with renal dysfunction. Bivalirudin is an excellent alternative to unfractionated or low-molecular-weight heparin (LMWH) in patients with a history of heparin-induced thrombocytopenia. It is at least equivalent to heparin plus a glycoprotein IIb/IIIa inhibitor in reducing ischemic events associated with UA and/or NSTEMI, with the added benefit of a reduction in bleeding.46 Up until recently, the role of bivalirudin in STEMI was uncertain. The HORIZONS-AMI trial randomized 3602 patients with STEMI undergoing primary PCI to UFH plus a glycoprotein IIb/IIIa inhibitor or to bivalirudin alone (with provisional glycoprotein IIb/IIIa in the cardiac catheterization lab).47 Major adverse cardiac event (MACE) rates were equivalent, but use of bivalirudin alone was associated with a 40% reduction in bleeding (4.9% versus 8.3%, P < 0.001;). However, at 1 year, MACE rates were similar in the two groups (11.9% versus 11.9%, HR 1.00, 0.82-1.21, P = 0.98), but there was a decrease in all-cause mortality with bivalirudin (3.4% versus 4.8%, P = 0.03).48
Enoxaparin is an LMWH with established efficacy as an anticoagulant in patients with STEMI who have received fibrinolytics or are undergoing PCI.49,50 The standard dose of enoxaparin is a 30-mg intravenous (IV) bolus, followed 15 minutes later by subcutaneous injections of 1 mg/kg every 12 hours. Patients with decreased creatinine clearance or older than 75 are at higher risk of bleeding with standard-dose enoxaparin and should not receive a bolus, but can receive a reduced dose of 0.75 mg/kg every 12 hours. Patients undergoing PCI should have an additional bolus if the last dose was given 8 to 12 hours prior. Maintenance dosing of enoxaparin should be given during the hospitalization (up to 8 days).
Fondaparinux, also an LMWH, can be dosed daily in patients receiving fibrinolytics for STEMI (initial dose of 2.5 mg IV followed by subcutaneous injections of 2.5 mg once daily). The OASIS-6 trial randomized over 12,000 patients with STEMI to 2.5 mg of fondaparinux or placebo. Death or reinfarction at 30 days was significantly reduced in the fondaparinux group (9.7% versus 11.2%, P = 0.008) and were maintained at 6 months.51 Severe bleeds were reduced with fondaparinux (61 versus 79, P = 0.13), and significant benefit was seen in patients who received fibrinolytics, as well those who were not reperfused. However, in patients undergoing PCI for STEMI, fondaparinux should not be administered alone, owing to an increased rate of catheter-related thrombosis observed in clinical trials.51,52 If fondaparinux has been chosen, unfractionated heparin should be administered with fondaparinux in the catheterization laboratory. Table 76-1 summarizes typical antiplatelet and anticoagulant therapy for ACSs.
TABLE 76-1 Antiplatelet/Anticoagulant Therapy in Acute Coronary Syndromes
| Drug | Initial Medical Treatment |
|---|---|
| Antiplatelet Drugs | |
| Aspirin | 162 to 325 mg nonenteric formulation, orally or chewed |
| Clopidogrel Prasugrel | LD of 300 to 600 mg orally, MD of 75 mg orally per day LD of 60 mg orally, MD of 10 mg orally per day |
| Ticlopidine | LD of 500 mg orally, MD of 250 mg orally twice daily |
| Anticoagulants | |
| Unfractionated heparin | LD of 60 U per kg (max 4,000 U) as IV bolus MD of IV infusion of 12 U/kg/h (max 1000 U/h) to maintain APTT at 1.5 to 2.0 times control (approximately 50-70 sec) |
| Enoxaparin | LD of 30 mg IV bolus may be given MD of 1 mg/kg subcutaneously every 12 h; extend dosing interval to 1 mg/kg every 24 h if estimated creatinine clearance <30 mL/min |
| Fondaparinux | 2.5 mg subcutaneously once daily. Avoid for creatinine clearance <30 mL/min |
| Eptifibatide | LD of IV bolus of 180 µg/kg MD of IV infusion of 2 µg/kg/min; reduce infusion by 50% in patients with estimated creatinine clearance <50 mL/min |
| Tirofiban | LD of IV infusion of 0.4 µg/g/min for 30 min MD of IV infusion of 0.1 µg/kg/min; reduce rate of infusion by 50% in patients with estimated creatinine clearance <30 mL/min |
| Bivalirudin | 0.1 mg per kg bolus, 0.25 mg/kg/h infusion |
LD, loading dose; MD, maintenance dose.
Adapted from Anderson JL, Adams CD, Antam EM et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non–ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2007;50:e1-157.
Nitrates
Nitrates have a number of beneficial effects in AMI. They reduce myocardial oxygen demand by decreasing preload and afterload, and they may also improve myocardial oxygen supply by increasing subendocardial perfusion and collateral blood flow to the ischemic region.53 Occasional patients with ST elevation due to occlusive coronary artery spasm may have dramatic resolution of ischemia with nitrates. In addition to their hemodynamic effects, nitrates also reduce platelet aggregation. Despite these benefits, the GISSI-3 and ISIS-4 trials failed to show a significant reduction in mortality from routine acute and chronic nitrate therapy.54,55 Nonetheless, nitrates are still first-line agents for the symptomatic relief of angina pectoris and when MI is complicated by congestive heart failure.
Beta-Blockers
Beta-blockers are beneficial both in the early management of MI and as long-term therapy. In the prefibrinolytic era, early IV atenolol was shown to significantly reduce reinfarction, cardiac arrest, cardiac rupture, and death.56 In conjunction with fibrinolytic therapy with tPA, immediate β-blockade with metoprolol resulted in a significant reduction in recurrent ischemia and reinfarction, although mortality was not decreased.57
The COMMIT-CCS 2 trial of 45,852 patients with acute MI had a factorial arm (the clopidogrel arm was discussed earlier) and randomized patients—93% of whom had STEMI and 54% of whom were treated with lytics—to treatment with metoprolol (3 IV injections of 5 mg each followed by oral 200 mg/day for up to 4 weeks) or placebo.58 Surprisingly, there was no difference in the primary endpoint of death, reinfarction, or cardiac arrest by treatment group (9.4% for metoprolol versus 9.9% for placebo, P = NS) or in the co-primary endpoint of all-cause mortality by hospital discharge (7.7% versus 7.8%, P = NS). Although reinfarction was lower in the metoprolol group (2.0% versus 2.5%, P = 0.001). there was an increase in the risk of developing heart failure and cardiogenic shock (5.0% versus 3.9%, P < 0.0001).58 Death due to shock occurred more frequently in the metoprolol group (2.2%, versus 1.7%), while death due to arrhythmia occurred less frequently in the metoprolol group (1.7%, n = 388 versus 2.2%, n = 498). Based on these findings, routine use of intravenous beta-blockers in the absence of systemic hypertension is no longer recommended.59
In contrast to the use of early aggressive beta-blocker therapy, the long-term use of beta-blockers post MI has favorable outcomes on mortality.60,61 The CArvedilol Post-infaRct survIval COntRolled evaluatioN (CAPRICORN) trial was a randomized placebo-controlled trial designed to test the long-term efficacy of carvedilol on morbidity and mortality in patients with LV dysfunction 3 to 21 days after MI who were already treated with angiotensin-converting enzyme (ACE) inhibitors.62 After an average follow-up period of 1.3 years, cardiovascular mortality was lower in the carvedilol arm (11% versus 14% for placebo, P = 0.024), as was all-cause mortality or nonfatal MI (14% versus 20%, P = 0.002).62 This study supports the claim that beta-blocker therapy after acute MI reduces mortality irrespective of reperfusion therapy or ACE inhibitor use. Relative contraindications to oral beta-blockers include heart rate less than 60 bpm, systolic arterial pressure less than 100 mm Hg, moderate or severe LV failure, signs of peripheral hypoperfusion, shock, PR interval greater than 0.24 second, second- or third-degree AV block, active asthma, or reactive airway disease.59
Angiotensin-Converting Enzyme Inhibitors
ACE inhibitors have been shown unequivocally to improve hemodynamics, functional capacity and symptoms, and survival in patients with chronic congestive heart failure.63,64 Moreover, ACE inhibitors prevent the development of congestive heart failure in patients with asymptomatic LV dysfunction.65 This information was the spur for trials evaluating the benefit the prophylactic administration of ACE inhibitors in the post-MI period. The SAVE trial showed that patients with LV dysfunction (LVEF < 40%) after MI had a 21% improvement in survival after treatment with the ACE inhibitor, captopril.66 A smaller but still significant reduction in mortality was seen when all patients were treated with captopril in the ISIS-4 study.55 The HOPE trial randomized 9297 patients with documented vascular disease or those at high risk for atherosclerosis (diabetes plus at least one other risk factor) in the absence of heart failure to treatment with the tissue-selective ACE inhibitor, ramipril (target dose 10 mg/day), or placebo.67 An impressive 22% reduction in the combined endpoint of cardiovascular death, MI, and stroke was observed, and the improved survival was additive to the benefits of aspirin and beta-blockers.67 The mechanisms responsible for the benefits of ACE inhibitors probably include limitation in the progressive LV dysfunction and enlargement (remodeling) that often occur after infarction, but a reduction in ischemic events was seen as well.
Immediate IV ACE inhibition with enalaprilat has not been shown to be beneficial,68 but oral ACE inhibition should be started early in the hospital course. Patients should be started on low doses of oral agents (captopril, 6.25 mg three times daily) and rapidly increased to the range demonstrated beneficial in clinical trials (captopril, 50 mg three times daily; enalapril, 10-20 mg twice daily; lisinopril, 10-20 mg once daily; or ramipril, 10 mg once daily).



Full access? Get Clinical Tree


