Chapter 84 Acid–base balance and disorders
THEORETICAL CONSIDERATIONS
The structural integrity of intracellular enzymes is essential for survival. Proton activity at enzymatic sites of action in cytosol and organelles must be tightly controlled. In critical illness, with survival under threat, direct monitoring of any intracellular site remains an impractical ideal. Clinicians are obliged to track extracellular data, usually from tests on arterial blood, knowing that plasma pH exceeds intracellular pH by 0.6 pH units on average.
WATER DISSOCIATION AND ACID–BASE
Stewart reminded us of the central role of water in aqueous acid–base equilibria.1 Mammals are approximately 60% water. It follows that the behaviour of water is fundamental to our understanding of acid–base physiology. Water (simplistically) dissociates as follows:
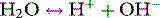
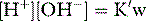
THE PaCO2/PH RELATIONSHIP – THE ACID–BASE ‘WINDOW’ FOR CLINICIANS
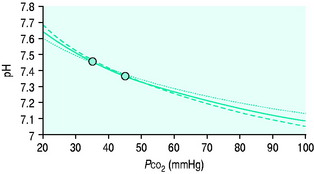
Clinicians use the relationship between arterial PCO2 (PaCO2) and arterial pH as the acid–base assessment platform. This is appropriate, because the PaCO2/pH curve is a fundamental physiological property (Figure 84.1). Several factors determine the shape and position of this curve.
THE PaCO2/pH RELATIONSHIP IS DEFINED BY SEVERAL SIMULTANEOUS EQUATIONS
Therefore apart from Equation 1, several other equations must be satisfied simultaneously at any equilibrium. They relate to:
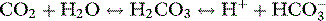
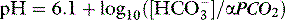
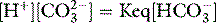
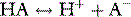
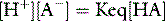
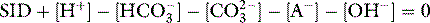
The trapped anions have weak acid properties. Any pH shift alters their negative charge, driving further ionic redistributions, particularly chloride, between compartments. The net effect is that plasma SID goes up and down with PaCO2 (Figure 84.2), the origin of the so-called Hamburger effect. Importantly, ionic shifts are confined within the total extracellular space, so that extracellular SID does not alter with PCO2. This is fortuitous for clinicians, forming the basis of the CO2 invariance of standard base excess (see below).
Thus it can be seen that for any individual the PaCO2/pH relationship is a unique acid–base ‘signature’ (Figure 84.1) and ultimately a complex function of extracellular SID and ATOT.
WEAK IONS AND BUFFER BASE
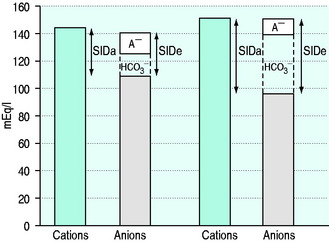
SID is a charge space. Weak ions, which arise from variably dissociating conjugate bases, occupy this space. These include H+, OH−, HCO3−, CO32− and A−. Their total net charge must always equal SID. However, HCO3− and A−, together known as the ‘buffer base’ anions, take up virtually the entire space on their own (Figure 84.2) since the other ions are measured in either micromoles/l or, in the case of protons, nanomoles/l. SID therefore not only dictates the buffer base concentration but is also numerically identical to it. In other words, SID = [HCO3−] + [A−]. This fact, plus Figge’s linear approximations2 for calculating A−, allows us to simplify Stewart’s equations in plasma, reducing them to three without sacrificing accuracy.3
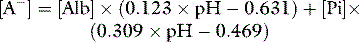
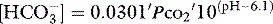
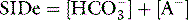
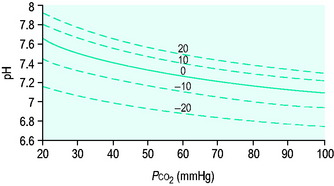
[Alb] is albumin concentration expressed in g/l. [Pi] is phosphate concentration in mmol/l. PCO2 is in mmHg. SIDe is effective SID, also known as ‘buffer base’ (Figure 84.2).
SID calculated from measured plasma concentrations of strong ions is termed the ‘apparent’ SID, or SIDa (Figure 84.2). Discrepancies between SIDe and SIDa imply the presence of unmeasured ions in plasma (see below).
ISOLATED CHANGES IN SID AND ATOT
At any given PaCO2, a falling SID or a rising ATOT reduce pH, moving the equilibrium towards a metabolic acidosis. Conversely, a rising SID or a falling ATOT create a metabolic alkalosis. Many argue that SID and ATOT act individually and should therefore be regarded as independent metabolic acid–base variables. By this argument we could have a strong ion acidosis or alkalosis combined with either a hyperalbuminaemic (high ATOT) acidosis or a hypoalbuminaemic (low ATOT) alkalosis.4 However, SID and ATOT do appear to be linked, with the SID set-point adjusting to ATOT. In particular, SID seems to fall in hypoalbuminaemia, presumably by renal chloride adjustment.5,6
HOW ACID–BASE DISTURBANCES AFFECT THE PACO2/PH RELATIONSHIP
Acute respiratory disturbances move data points along the prevailing PaCO2/pH curve, to the left in respiratory alkalosis, and to the right in respiratory acidosis (Figure 84.1). In contrast, metabolic disturbances (altered extracellular SID and/or ATOT) shift the entire curve up or down (Figure 84.3). A down-shifted curve means that the pH at any given PaCO2 is lower than normal, which – depending on the PaCO2 – represents either a primary metabolic acidosis or else metabolic compensation for a respiratory alkalosis. With an up-shifted curve, the pH at any given PaCO2 is higher than normal, signifying either a primary metabolic alkalosis or else compensation for a respiratory acidosis.
TEMPERATURE CORRECTION OF BLOOD GAS DATA – ‘ALPHA-STAT’ VERSUS ‘PH-STAT’ APPROACHES
Blood gas analysers operate at 37 °C. Their software can convert pH and gas tensions to values corresponding to the patient core temperature for interpretation and action. This is the ‘pH-stat’ approach. The alternative isto act on values as measured at 37 °C – the ‘alpha-stat’ approach.
Hence, a simple way to keep alpha at 0.55 in hypothermia is to maintain uncorrected PaCO2 and pH measurements in their 37 °C reference ranges.7,8 This mimics the hypothermic physiology of ectothermic (cold-blooded) animals. Similar arguments apply in fever, the more common intensive care unit (ICU) scenario. Many intensivists follow the alpha-stat approach, whatever the core temperature.
Those who favour the pH-stat approach argue that it is more consistent with the physiology of hibernating endothermic mammals, and that it allows better maintenance of cerebral perfusion in hypothermia.9 This approach was used during an influential trial of mild hypothermia following out-of-hospital cardiac arrest.10
RENAL PARTICIPATION IN ACID–BASE
In the absence of renal function, there is a progressive metabolic acidosis. About 60 mEq of strong anions, particularly sulphate, but also hippurate and others, are produced daily as metabolic end-products. These accumulate in renal failure, reducing extracellular SID. So does free water, which brings sodium concentrations closer to chloride, again reducing SID. Hyperphosphataemia contributes by increasing ATOT, although in acute renal failure this is commonly offset by coexistent hypoalbuminaemia.11
Traditionally, renal acid–base homeostasis is described in terms of resorption of filtered bicarbonate primarily in the proximal tubule, and excretion of fixed acids through titration of urinary buffers, particularly phosphate, and through excretion of ammonium, primarily in the distal tubule.12
From the physical chemical perspective, the traditional analysis of renal acid–base homeostasis is misleading, since it is based on H+ or HCO3− ‘balances’. H+ and HCO3− are dependent variables, responsive exclusively to PCO2, SID and ATOT, and not subject to ‘in versus out’ balance sheets. The physical chemical explanation is simple. The kidneys regulate extracellular SID via urinary SID, the principal tool being tubular NH4+ acting as an adjustable cationic partner for tubular Cl− and other urinary strong anions.13 The kidneys also modify ATOT via phosphate excretion, which is a totally different concept from that of ‘titratable acidity’.
ACID–BASE ASSESSMENT – THE TWO ‘SCHOOLS’
By convention, acid–base disorders are divided into respiratory (PaCO2) and metabolic (non-PaCO2). PaCO2 is the undisputed index of respiratory acid–base status. Two ‘schools’, Boston and Copenhagen, separated by a large ocean,14 have formed around the identification and quantification of metabolic acid–base disturbances. Both succeed as navigation systems, if used correctly.
Stewart’s concepts neither invalidate nor supplant the traditional approaches,15–17 but rather help us to understand their physiological basis, evaluate their relative merits, and extend their utility.18 SID by itself is not a reliable measure of metabolic acid–base status, for three reasons:
BASE EXCESS AND STANDARD BASE EXCESS
In 1960, Siggaard-Andersen introduced ‘base excess’ (BE).19 BE was defined as zero when pH = 7.4, PCO2 = 40 mmHg (both at 37 °C). If pH ≠ 7.4 or PCO2 ≠ 40 mmHg, BE was defined as the concentration of titratable hydrogen ion required to return the pH to 7.4 while maintaining PCO2 at 40 mmHg.
In the lead-up, Astrup, Siggaard-Andersen, Engel and others had equilibrated the blood of Danish volunteers with known CO2 tensions at varying haemoglobin concentrations, after first adding known amounts of acid or base. The data were then used to create an ‘alignment nomogram’ which allowed the determination of BE from simultaneous measurements of pH, PCO2 and haemoglobin concentration.
Seventeen years later, Siggaard-Andersen published the Van Slyke equation for calculating BE.20 It was derived from known physical chemical relationships, and was claimed to match the empiric nomogram. The Van Slyke equation computes (Δ[HCO3−] + Δ[A−]) – in other words, the deviation from normal of the buffer base concentration in whole blood. From the Stewart perspective, buffer base and SID are interchangeable terms. Hence Stewart would describe BE as the abnormality in whole blood SID at the prevailing ATOT.
It became clear that BE loses its CO2 invariance in vivo, where Gibbs Donnan forces drive ions between intravascular and interstitial compartments. As a result, a primary change in PaCO2 shifts BE in the opposite direction. The solution was to calculate BE at a haemoglobin concentration of approximately 50 g/l, replicating the mean extracellular haemoglobin concentration and thus more closely modelling the extracellular environment.21 This is standard base excess (SBE).
As a metabolic acid–base index, SBE is close to ideal, being both quantitative and demonstrably independent of PacO2.22 A useful formula is:
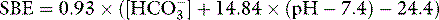
with SBE and [HCO3−] values in mEq/l. This formula can be further refined to allow for changes in plasma ATOT due to variations in albumin and phosphate.23 However, because haemoglobin is the predominant weak acid, the end result is very similar.
A typical SBE reference range (in mEq/l) is −3.0 to +3.0. If SBE < −3.0 mEq/l, there is a down-shifted PaCO2/pH curve. This could represent either a primary metabolic acidosis or else compensation for a primary respiratory alkalosis, depending on the PaCO2 and the pH (see below). SBE quantifies the increase in extracellular SID (in mEq/l) needed to shift the curve back to the normal position without changing ATOT (Figure 84.3). In terms of the original BE definition, this is roughly the required dose of sodium bicarbonate in mmol per litre of extracellular fluid. Similarly, if SBE is > 3.0 mEq/l, there is an up-shifted curve, either a metabolic alkalosis or compensation for a respiratory acidosis. The SBE is the decrease in extracellular SID needed to shift the curve back to the normal position at the prevailing ATOT. Conceptually, it approximates the dose of HCl required per litre of extracellular fluid. SBE is thus ‘extracellular SID excess’ or ‘SIDex’.24



Full access? Get Clinical Tree


