Fig. 44.1
Historical overview of influence of pharmacogenetics on anesthesia.βARs β-adrenergic receptors,µ-OR µ-opioid receptor,SCN9A sodium channel mutation
Prolonged Apnea After Succinylcholine
The depolarizing muscle relaxant succinylcholine entered clinical practice in 1951. Paralysis from succinylcholine peaks in approximately 45 seconds and usually lasts for 2 to 6 minutes, convenient for facilitating tracheal intubation. In 1952, anesthesiologists reported rare cases of prolonged paralysis after its use. Biochemical studies revealed why. Plasma butyrylcholinesterase (BChE, also called pseudocholinesterase) hydrolyzes succinylcholine to inactive components. Succinylcholine acts quickly and briefly in individuals with normal BChE activity, but abnormal BChE activity in 1:2500 (0.04%) of Caucasians [8,9] prolongs succinylcholine activity’and paralysis’by 5 to 10-fold. In 1952, Bourneet al. [10] reported that “those patients who recover slowly from succinylcholine had significantly lower pseudocholinesterase levels than did patients recovering at the normal speed.” Lehmann and Evans, and Kalow independently established the mechanism. Lehmann’s group detected decreased levels of BChE in patients with prolonged paralysis [11], leading to their suggestion that a genetic cause, rather than chronic disease or chemical poisoning, altered BChE activity. In their 1956 landmark publication, Lehmann and Evans investigated families of 5 unrelated patients having prolonged responses to succinylcholine [12], finding otherwise healthy family members with low BChE activity without any reason for such levels. These findings provided a genetic link to prolonged paralysis after succinylcholine.
In 1951, Werner Kalow (Fig. 44.2) proved that procaine-esterase was actually BChE. He provided a more efficient method to measure esterase activity. Serendipitously, he contacted Donald Gunn, a physician using low-dose succinylcholine to dampen muscular contractions during electric shock therapy. Gunn noted that a few patients remained relaxed for almost an hour rather than 2 to 6 minutes. Kalow discovered that BChE enzymes from 2 of these patients behaved differently from normal BChE, and inferred that a genetic change caused a structural difference in the enzyme. In the families of these two patients, Kalow found differential inhibition of BChE by a local anesthetic called dibucaine (also known as cinchocaine or nupercaine). Dibucaine inhibited 79% of normal BChE activity. In both parents of the two patients, inhibition was only 62%, and minimal inhibition (16%) occurred in patients having a prolonged effect of succinylcholine. Such patients had two abnormal genes for BChE (homozygous trait); the parents were heterozygous with one normal and one abnormal gene, and the average person had two normal genes. Succinylcholine was one of the first drugs whose metabolism was shown to be affected by genes displaying simple Mendelian patterns of inheritance [13].

Fig. 44.2
Werner Kalow (1917–2008) joined the University of Toronto, Canada, in 1951, and remained active in academics as a pharmacologist until the year of his death. (From Kalow W: Atypical plasma cholinesterase. A personal discovery story: a tale of three cities. Can J Anaesth 2004; 51: 206–11, with permission.)
Kalow and his co-workers then demonstrated that individuals having a prolonged response to succinylcholine had abnormally functioning BChE rather than a low quantity of enzyme. They had a genetically determined defect in BChE’a so-called dibucaine resistant enzyme, or atypical enzyme’causing the abnormal reaction to succinylcholine. Kalow’s use of dibucaine inhibition percentage remains the standard for classifying heterozygous, homozygous, and wild-type phenotypes of BChE. His findings and his study of other examples of varied responses to drugs, motivated him to write his 1962 book “Pharmacogenetics: Heredity and the Response to Drugs” [7] establishing pharmacogenetics as a new scientific discipline. Subsequently, over 40 polymorphisms associated with BChE deficiency were described, the most common being the A and the K enzyme variants. The A (for “atypical”) variant has an altered activity while the K variant (after Kalow) results in a lesser production of enzyme. Although prolonged paralysis following succinylcholine is now a rare but easily managed clinical problem, the finding of prolonged muscle relaxation with succinylcholine in specific individuals provided the first evidence for serious clinical effects from genetic variation, and highlighted anesthesiology as a discipline wherein genetics had immediate clinical implications.
Malignant Hyperthermia
Succinylcholine contributed to another anesthesia-related pharmacogenetic discovery. Malignant hyperthermia (MH) is an uncommon adverse pharmacogenetic syndrome associated with administration of succinylcholine and/or inhaled volatile anesthetics [14]. The mechanism triggering this reaction remains unknown (severe stress alone can trigger MH). When triggered, MH increases cytosolic Ca2+ in skeletal muscle cells, and this increase can produce hyperthermia, hypermetabolism, muscle rigidity, breakdown of muscle tissue, and death [15].
Here is the story of the discovery of MH. On 8 April 1960, a car hit a 21 year old student, fracturing his right tibia and fibula [16]. Upon arrival at the casualty department of the Royal Melbourne Hospital, in Victoria, Australia, he and his mother expressed more concern about the anesthesia than his leg. Ten family members (3 cousins and 7 aunts and uncles), out of 38, had died during or shortly after receiving ether anesthesia. This family history caused the student’s mother to request a local anesthetic when the boy had an appendectomy at age 12. The consultant anesthesiologist on 8 April was James Villiers. Having heard the story, he and the surgeon learned that the patient’s cousin had recently been safely anesthetized with a “modern anesthetic” at the nearby children’s hospital. After excluding porphyria, the only condition known at the time to affect anesthesia, Villiers believed that the deaths must have been due to “ether convulsions” and recommended using a new, possibly safer anesthetic, halothane’plus vigilant monitoring. Ten minutes into anesthesia, hypotension, tachycardia, cyanosis, and hyperventilation occurred, and the soda lime canister became very hot. The fractures were quickly set, and anesthesia administration discontinued. The patient remained cyanotic and unresponsive and became hot and sweaty. He was immediately packed in ice from a nearby cardiac theater, and within an hour, he was awake and alert. His subsequent course was uneventful. He was the first recorded patient to have had’and survive’MH [17].
Villiers referred the patient to Richard Lovell, who, in turn asked his research assistant, Michael Denborough (Fig. 44.3), to see the patient. Denborough had an interest in genetics, and the familial history led him to suspect a previously unrecognized inborn error of metabolism. When the student again required anesthesia for an impacted renal calculus in 1961, John Forster of the Royal Melbourne Hospital gave him an uneventful spinal anesthetic [16].

Fig. 44.3
(left) and James Villiers (right), photographed in 2012. (Courtesy of Christine Ball)
In 1964, deaths were noted to have occurred during anesthesia in a large Wisconsin (USA) family. One child had died in Toronto, Canada, and Beverly Britt, the anesthesiologist, along with Kalow and RA Gordon, a professor of anesthesia, organized inquiries throughout Canada. This led to presentation of 13 cases of MH at a 1966 symposium in Toronto [18]. These patients had evinced two common features: muscle rigidity and high temperature. The first description in the American literature of a patient with MH, with reported blood gas data, was similar to that of Villiers in that the patient survived after being packed in ice [19]. When this case was presented at the annual American Medical Association meeting, several in the audience reported caring for similar patients who had received halothane and succinylcholine.
In November 1969, a patient at the Royal Melbourne Hospital received halothane and succinylcholine and died from MH [20]. This patient’s right arm had became rigid, prompting a test of plasma creatine kinase activity, an enzyme involved in energy production in muscle fibers. Creatine kinase activity was increased, indicating muscle breakdown. The original student who had broken his leg in 1960, and some of his relatives also were found to have increased serum creatine kinase activity’evidence of a generalized muscular disease [21]. In 1970, in Johannesburg South Africa, H Isaacs and M Barlow independently observed increased serum creatine kinase levels in relatives of MH patients [22]. Impressed by the muscle rigidity, Kalow and Britt tested their muscle samples for contractile forcein vitro. Muscle from patients with MH exhibited abnormally strong contractions in the presence of caffeine. Caffeine was used because it inducesin vitro skeletal muscle contraction. This resulted in the first clinically useful test for detection of a predisposition to MH in 1970 [23]. In 1971, F Ellis and colleagues showed that halothane augments muscle contraction in patients who are susceptible to MH [24].
In the early 2000s, molecular genetic testing for MH susceptibility was introduced in Europe [25] and North America [14]. The genetic assessments were based on DNA sequence variations in the skeletal muscle type ryanodine receptor (RYR1) gene, which encodes a protein involved in the excitation-contraction process of muscle cells. In 1990, MacLennanet al. had identified theRYR1 gene as a candidate gene that might mediate MH [26]. More than 180RYR1 variants have since been documented, with 29 considered mutations predisposing to MH [27,28]. In 2001, the European Malignant Hyperthermia Group published criteria for proof of causation, and agreed on guidelines for molecular genetic detection for susceptibility to MH. These guidelines are the first to describe genetic screening for testing in anesthesiology [25,29,30].
CYP2D6 Enzyme Variation
The next pharmacogenetic discovery relevant to anesthesia came in the late 1960s, in association with a description of an enzyme, now called CYP2D6. CYP2D6 affects metabolism of 25% of the drugs used in medicine, including anesthesiology [31]. CYP2D6 is part of a family of cytochrome P450 (CYP) drug metabolizing enzymes, enzymes involved in metabolism of morphine derivatives, β-blockers, antiarrhythmics, antipsychotics, and antiemetic drugs [31]. Polymorphisms (specific variations in protein structure and function) inCYP2D6 can affect drug efficacy, side effects, and toxicity. Beginning in 1967, 3 laboratories independently discovered polymorphic CYP2D6.
Folk Sjöqvists’ group in Sweden found that metabolism of two CYP2D6 substrates showed considerable inter-individual variation in metabolite plasma levels. Two phenotypes were identified in 1967 [32]. Studies in 19 identical (monozygotic) and 20 fraternal (dizygotic) sets of twins in 1969, suggested the difference was gene-based [33].
At St. Mary’s Medical School in London in 1975, Robert Smith studied debrisoquine, a new agent for treating hypertension. Several healthy volunteers, including Smith, received 40 mg of debrisoquine. Everyone but Smith seemed unfazed by the injection, but Smith became severely hypotensive and collapsed. He recovered fully, but his startling reaction prompted further investigation, revealing that his debrisoquine hydroxylase, the enzyme that metabolizes debrisoquine, eliminated only 10% of the drug. The normal enzyme eliminates 80%. Smith then identified 2 medical students with deficient debrisoquine hydroxylase, traced the family histories, and found that the trait was inherited. His team then tested hundreds of volunteers, discovering that 1 in 10 developed hypotension after debrisoquine. Although 10% of the population were homozygous carriers, 45% were heterozygote carriers of the deficient gene and could transmit it to their children. Smith’s observations had uncovered a mechanism explaining numerous adverse drug reactions [34].
Finally, Michel Eichelbaum’s group in Bonn, Germany, studied the antiarrhythmic effects of sparteine [35]. Two subjects complained of double vision, blurred vision, dizziness, and headache after receiving the drug. Both had 4–5 times higher sparteine plasma levels than other individuals, demonstrating a defect in the capacity to oxidize some drugs, a defect inherited as an autosomal recessive trait. In 1980, Bertilssonet al. in Sweden showed that subjects having an impaired metabolism of debrisoquine, also had an impaired metabolism of sparteine [36]. Also in 1980, Kalow found that students of Europeanversus Chinese descent differed in their capacity to metabolize debrisoquine and sparteine [37], the first observation of inter-ethnic differences in drug metabolism. Debrisoquine and sparteine metabolism were suspected to be genetically linked [38]. A search then began for the gene underlying their variable pharmacokinetics.
In 1985, Distlerathet al. purified the protein’CYP2D6’responsible for the altered metabolism from human liver microsomes [39]. In 1987, Eichelbaumet al. localized the gene encoding CYP2D6 to chromosome 22 [40]. CYP2D6 was the first polymorphic P450 to be characterized at a molecular level, initially identified as a gene responsible for altered metabolism of debrisoquine, sparteine, and other drugs [41]. Further study revealed that CYP2D6 is the only non-inducible enzyme of the P450 family of enzymes. Non-inducible means that it is constantly expressed rather than being turned on or off. As a result, a mutation of the enzyme may cause sustained inter-individual variation in enzyme activity [31]. Genotyping for CYP2D6 activity is drug specific, and the need for typing an individual is limited. If genotyping is needed, patients are assigned to one of four categories based on their ability to metabolize CYP2D6 substrates: ultrarapid metabolizers (UM), extensive metabolizers (EM), intermediate metabolizers (IM), and poor metabolizers (PM) [42]. PMs (two non-functional gene alleles) metabolize CYP2D6 substrates less well than their counterparts (IMs, EMs, UMs). UMs possess multiple functional copies of the same normal CYP2D6 gene, and an increase in the number of functional copies in an individual increases the rate of metabolism of CYP2D6 substrates (Fig. 44.4).
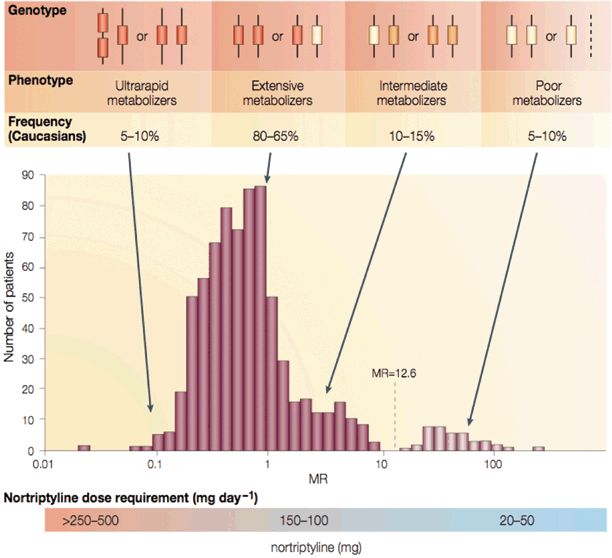
Fig. 44.4
CYP2D6 activity predicts ability to metabolize a wide range of drugs (see text for details). Clinical doses of nortriptyline (a tryicyclic antidepressant) to be used in different genotypes are indicated. (From Meyer UA: Pharmacogenetics’five decades of therapeutic lessons from genetic diversity. Nat Rev Genet 2004; 5: 669–76 with permission.)
CYP2D6 genetic variations have other clinical implications. In 2006, Korenet al. [43] described a neonatal death leading to the suggestion that breastfeeding mothers taking codeine for postpartum pain should be genotyped for CYP2D6 activity. CYP2D6 O-demethylates codeine to morphine. Mothers and neonates that are CYP2D6 UMs or EMs may produce toxic blood levels of morphine or its active metabolite morphine-6-glucuronide (M6G). The infant in this case report was a CYP2D6 EM, and had a blood concentration of 70 ng/ml morphine; neonates breastfed by mothers receiving codeine normally have concentrations of 0–2.2 ng/ml [43]. The mother was a CYP2D6 UM, and her breast milk had 87 ng/ml morphine’the normal range being 1.9–20.5 ng/ml at doses of 60 mg codeine every 6 hours [43]. Thus the infant had 2 reasons for increased morphine levels. These findings and those from case-control studies [44] indicate that codeine should be avoided in breastfeeding mothers with CYP2D6 EM and UM genotypes.
Genetics and Pharmacodynamic Variation
Pharmacokinetics (what the body does to a drug) and pharmacodynamics (what a drug does to the body) can influence the effectiveness and metabolism of medicines given to produce anesthesia. The previous section concentrated on pharmacokinetics, identifying genetic alterations in drug metabolizing enzymes that change circulating drug levels. The next section explores pharmacodynamic effects, specifically genetic variations in receptor proteins that may alter activation of signaling pathways, despite the presence of constant drug concentration. We present 3 clinically relevant examples.
β-Adrenergic Receptors and Altered Physiologic Responsiveness
β-adrenergic receptors (βARs) influence cardiac contractility, heart rate, and vessel dilation. Anesthesiologists use βAR agonists and antagonists to ensure intraoperative hemodynamic stability and improve perioperative outcome [45]. β2AR genetic variants, generally single nucleotide polymorphisms (SNPs), were initially identified in 1993 and 1994, and clinically relevant β1AR SNPs were identified in 1999 and 2000 [46]. β1AR and β2AR antagonist therapy after acute myocardial infarction (MI) decreasesaggregate patient mortality, a fact known for decades [47]. In 2005 Lanfearet al. [48] asked whether specific sub-groups of individuals benefited more than others, from βAR antagonist therapy. Examining four common β1 and β2AR SNPs, Lanfear foundincreased mortality with βAR blocker therapy in individuals with genetic variants in the gene encoding the β2AR (ADRB2) (about 25% of the acute MI population), whereas the remaining patients benefited. Such findings suggest that β2AR DNA testing might indicate which patients should receive βAR antagonists after MI. If these findings are reproduced, such genetic testing would routinely precede the use of βAR antagonists after MI.
Airway hyper-reactivity associated with asthma is a concern to anesthesiologists. Bronchodilators prescribed to prevent or to treat airway hyper-reactivity interact with β2ARs. In 1999, Limaet al. [49] reported that asthmatic subjects who were homozygous for the Arg16 wild-type (normal) allele inADRB2 (i.e., they had 2 copies of the normal gene), initially demonstrated enhanced responsiveness to β2-agonist bronchodilators, compared to patients homozygous for the Gly16 variant. However, this acute protective effect of Arg16 differed from results in studies assessing prolonged β-agonist therapy. In 2004, Israelet al. [50] showed that mildly asthmatic patients who were homozygous for Gly16, improved with long-term albuterol therapy, a result subsequently confirmed [51]. Our current understanding is that in Arg16 homozygous patients, long-acting β-agonist therapy may increase severe asthma exacerbations. Alternative treatment strategies may better help this subgroup of individuals [52]. This provides another example of how pharmacogenetics may help guide and/or facilitate alternative treatment strategies [53].
µ-Opioid Receptor Variation and Pain Medicine Effectiveness
Opium has been harvested from poppies and smoked to relieve pain and alter mentation for centuries. Morphine, codeine, and non-narcotic alkaloids come from this folk remedy. Opium is also the precursor for recreational drugs such as heroin. Morphine and other clinically relevant opioid analgesics primarily target the µ-opioid receptor (µOR), a receptor whose gene,OPRM1, varies extensively. In May of 1998, Wendel and Hoehe described the µOR gene (OPRM1) [54]. Three months later, Bond and her colleagues discovered a SNP at position 118 in 10% of their study population, and suggested that this A118G polymorphism (in which guanine substitutes for adenine at position 118 in the receptor protein) alters the extracellular binding site of the µOR, thereby changing the binding affinity of β-endorphins (the natural endogenous biochemical for the opioid receptors) as well as exogenous opioids [55]. Two reports of the functional consequence of the A118G genetic variant came in 2002. Wandet al. [56] found that healthy volunteers carrying the G118 variant, had increased cortisol responses when given the opioid-antagonist, naloxone. Days later, Lötschet al. [57,58] suggested that the A118G polymorphism protects against morphine-6-glucuronide toxicity.
Variants of the µOR gene may explain inter-individual responses to opioid analgesia [59]. An approach to examining the heritable component of pain perception and response to opioids has historically involved twin studies [60]. Other clinical studies relevant to anesthesia have examined the potential pharmacogenetic effect of the common A118G polymorphism, on the response to various opioids after surgery, or when given to women for labor pain during epidural analgesia. Importantly, the dose of intravenous morphine required by post-surgical patients has been shown to vary, based on an individual’s µOR genotype [61]. Defining µOR genetic variants is needed to understand complex pain pathways, stratify pain phenotypes into their individual components, and create novel predictive genetic tests for use in pain medicine [62].
Sodium Channel Genetic Variants and Channelopathies
A rare, fascinating, and deadly condition preventing the perception of pain has enhanced our understanding of sodium channels’ role in pain perception. It has also entered popular culture (see “The Girl Who Played with Fire” by Stieg Larsson). Survival and protection of the body demands the perception of pain (e.g., to remove one’s fingers from a hot object). In 2006, Coxet al. [63] described the genetic basis of a congenital inability to experience pain. In unrelated consanguineous Pakistani families, Cox and colleagues identified 3 loss-of-function mutations ofSCN9A, a gene encoding for the alpha subunit of the voltage-gated Nav1.7 sodium channel, which, along with other voltage-gated sodium channels, subserves the generation and conduction of action potentials that mediate pain. Isolated patients and families subsequently demonstrated similar mutations [64–66]. Presently 15 loss-of-function recessive trait mutations of the channelopathy-associated insensitivity to pain phenotype are known [66].
Genetic and Genomic Methods, Tools, and Their Impact
Variation in response to disease onset and severity, or to drug treatments, may result from augmenting/dampening of cascades of genes. Animal models have facilitated examination of the effect of genetic variability in the context of a whole organism. We now review some of the most common of these non-human models to see how they have advanced the field.
Classic Non-Human Models
The study of genetics began with Mendel’s observations of pea plants. Animal models’rats, cats, dogs, and monkeys’have also been used. The most commonly used models today include the nematode Caenorhabditis elegans (C.elegans), the fruitfly Drosophila melanogaster (D.melanogaster), and genetically engineered mice (knock-out and transgenic mice). These models reflect key discoveries in medicine, as acknowledged by several Nobel Prizes. Below we summarize advances related to the understanding of genetics and anesthetic mechanisms for each of these classic genetic models.
C.elegans is a transparent roundworm approximately 1 mm long. It reproduces rapidly allowing generations to be studied quickly. This simple organism was the first multicellular organism with a nervous system to have its genome completely sequenced. Under normal conditionsC. elegans is almost constantly moving. When exposed to a volatile anesthetic, after an initial phase of increased locomotion, uncoordinated motion progresses to immobility. WhenC. elegans is removed from the anesthetic, motion returns quickly. Phil Morgan and Helmut Cascorbi first described this model as a tool to study the mechanism of action of volatile anesthetics in 1985 [67]. Over the last two decades, Mike Crowder, Phil Morgan, and Marge Sedensky applied this model to study those mechanisms [68]. The two most common endpoints used to measure anesthetic effects inC. elegans have been uncoordination (or change in motion caused by anesthetic exposure), and immobility. The mechanisms of the two anesthetic actions are distinct and most likely reflect different aspects of the complex behavioral effects.
Mutations in neuronal proteins (called syntaxin) ofC elegans significantly alter the sensitivity to volatile anesthetics (a 33-fold range of sensitivity for isoflurane), suggesting that syntaxin is a possible target of volatile anesthetics. Overall, genetic studies suggest that a vast group of molecular targets or pathways determine anesthetic sensitivity. More recent studies have identified additional sites of action of volatile anesthetics, including ion channels and mitochondrial proteins. The implication that mitochondria were involved in anesthetic sensitivity was highlighted by the observation that children anesthetized with sevoflurane to perform muscle biopsies for the diagnosis of suspected mitochondrial defects, had different anesthetic responses as a function of their mitochondrial function. Modification of genes have been used to create anesthesia sensitive and resistant worms, facilitating exploration of mechanisms of anesthetic action [68], such as identifying most recently that complex I of the electron transport chains (ETC) is a possible target of volatile anesthetics [69].
Physical characteristics of D.melanogaster such as eye bristles, wing patterns, antenna, and patterns on the head and thorax have been used as phenotypic markers for alterations in specific genes. As with C.elegans, the entire genome and several variants of D.melanogaster have been sequenced in the last decade. Howard Nash at the NIH, discovered strains of D.melanogaster that were either sensitive or resistant to inhaled anesthetics [70]. Genes that regulate states of arousal, gene copy number, and unusual ion channels that serve as a depolarizing leak may underlie resistance and sensitivity. The D.melanogaster nervous system stays active during anesthesia and in this, and overall conservation of genes and their function, provides parallels with the human nervous system.
Some of the yet unresolved questions about the mechanism of action of anesthesia are related to the fact that (1) the numerous endpoints associated with the anesthetic state are probably the result of effects at different sites of anesthetic action, (2) ‘immobility’ as observed in nematodes doesn’t reflect the state of ‘loss of consciousness and perception of pain’ that is the goal of anesthesia, and (3) genetic studies do not explore all the pathways involved in mechanism of action, such as regulation that occurs downstream.
Mario Capecchi, Martin Evans and Oliver Smithies received the Nobel Prize in Medicine for their 1989 creation of the first knockout mouse, a mouse lacking a specific gene. By inactivating a specific gene, and observing the consequent differences in physiologic expression and phenotype, the function of the gene can be inferred. These models have been used to further our understanding of human biology, including mechanisms of anesthetic action [71]. Such genetic manipulation allows a controlled change heretofore only available as natural variations as described above (e.g., consider the natural variations in the opioid receptor). Knockout mice have been used to assess the relevance of numerous ligand- and voltage-gated channels to mediate the effects of inhaled anesthetics, particularly their capacity to produce immobility in the face of noxious stimulation. However, despite high hopes, a review by Sonner in 2003 [72] and a follow-up review by Eger in 2008 [73] do not suggest that the hopes have been realized. Why? Knockout of a gene affects but one ion channel, and then only one member of what may be a large family. What if anesthesia results from actions on multiple channels? And the body may compensate for the loss of function produced by a knockout, obscuring the consequences of the knockout.
Related posts:
 History Reflected in the Evolving Approaches to Anesthesia for a Patient Undergoing Cholecystectomy
A History of Intravenous Anesthesia
History Reflected in the Evolving Approaches to Anesthesia for a Patient Undergoing Cholecystectomy
A History of Intravenous Anesthesia
 Major Anesthesia-Related Events in the 2000s and Beyond
Major Anesthesia-Related Events in the 2000s and Beyond
 1846–1860: Following the Discovery of Anesthesia
1846–1860: Following the Discovery of Anesthesia
 A History of Research in Anesthesia
A History of Research in Anesthesia
 The Evolution of Premedication
The Evolution of Premedication

Full access? Get Clinical Tree


