Edited by Anne-Maree Kelly Anne-Maree Kelly Headache is a common ailment that is often due to a combination of physical and psychological factors. The vast majority are benign and self-limiting and are managed by patients in the community. Only a very small proportion of patients experiencing headache attend emergency departments (ED) for treatment. The challenges are to distinguish potentially life-threatening causes from the more benign and to manage effectively the pain of headache. The structures in the head capable of producing headache are limited. They include: The bulk of the intracranial contents, including the parenchyma of the brain, the subarachnoid and pia mater and most of the dura mater, are incapable of producing painful stimuli. The pathological processes that may cause headache are: The pathophysiological causes of headache are summarized in Table 8.1.1. Table 8.1.1 A pathophysiological classification of headache In the assessment of a patient with headache, history is of prime importance. Specific information should be sought about the timing of the headache (in terms of both overall duration and speed of onset), the site and quality of the pain, relieving factors, the presence of associated features, such as nausea and vomiting, photophobia and alteration in mental state, medical and occupational history and drug use. Intensity of the pain is important from the viewpoint of management but is not a reliable indicator of the nature of underlying pathology. This said, sudden, severe headache and chronic, unremitting or progressive headache are more likely to have a serious cause. Physical examination should include temperature, pulse rate and blood pressure measurements, assessment of conscious state and neck stiffness and a neurological examination, including funduscopy (where indicated). Abnormal physical signs are uncommon, but the presence of neurological findings makes a serious cause probable. In addition, a search should be made for sinus, ear, mouth and neck pathology and muscular or superficial temporal artery tenderness. Some headaches have ‘classic’ clinical features: these are listed in Table 8.1.2. It must be remembered that, as with all diseases, there is a spectrum of presenting features and the absence of the classic features does not rule out a particular diagnosis. Every patient must be assessed on their merits and, if symptoms persist without reasonable explanation, further investigation should be undertaken. Table 8.1.2 Classic clinical complexes and cause of headache For the majority of patients with headache no investigation is required. The investigation of suspected subarachnoid haemorrhage and meningitis is discussed elsewhere in this book. If tumour is suspected, the investigations of choice are magnetic resonance imaging (MRI) or a contrast-enhanced computed tomography (CT) scan. An elevated erythrocyte sedimentation rate (ESR) may be supporting evidence for a diagnosis of temporal arteritis. With respect to sinusitis, facial X-rays are of very limited value. The pathological basis of tension headaches remains unclear, but increased tension of the neck or cranial muscles is a prominent feature. A family history of headaches is common and there is an association with an injury in childhood or adolescence. The most common precipitants are stress and alteration in sleep patterns. Aspirin, non-steroidal anti-inflammatory agents (NSAIDs) and paracetamol (acetaminophen) have all been shown to be effective in the treatment of tension headaches, with success rates between 50 and 70%. Ibruprofen 400 mg or ketoprofen 25–50 mg appear to be the most effective, followed by aspirin 600–1000 mg and paracetamol 1000 mg. Migraine can be a disabling condition for the sufferer. Most migraine headaches are successfully managed by the patient and their general practitioner, but a small number fail to respond or become ‘fixed’ and sufferers may present for treatment at EDs. As most patients (up to 80% in some studies) have tried oral medications prior to presenting, parenterally administered agents are usually indicated for ED treatment. Migraine is a clinical diagnosis and, in the ED setting, a diagnosis of exclusion. Other causes of severe headache, such as subarachnoid haemorrhage and meningitis, must be ruled out before this diagnosis is made. Of particular note, the response of a headache to antimigraine therapy should not be used to assume that the cause was migraine. There have been reports that the headaches associated with subarachnoid haemorrhage and meningitis have, on occasion, responded to these agents. The pathophysiology of migraine is complex and not completely understood. It is a chronic neurovascular disorder characterized by dysfunction of the central and peripheral nervous system and intracranial vasculature. The headache pain of migraine seems to result from the activation of the trigeminovascular system. The triggers for the development of migraine headache are probably chemical and are thought to originate in the brain, the blood vessel walls and the blood itself. These triggers stimulate trigeminovascular axons, causing pain and the release of vasoactive neuropeptides. These neuropeptides act on mast cells, endothelial cells and platelets, resulting in increased extracellular levels of arachidonate metabolites, amines, peptides and ions. These mediators and the resultant tissue injury lead to a prolongation of pain and hyperalgesia. Serotonin has also been specifically implicated in migraine. By activation of afferents, it causes a retrograde release of substance P. This in turn increases capillary permeability and oedema. Migraine is defined as an idiopathic recurring headache disorder with attacks that last 4–72 hours. Typical characteristics are unilateral location, pulsating quality, moderate or severe intensity and aggravation by routine physical activity. There is also usually nausea, photophobia and phonophobia. In some patients, migraine is preceded by an ‘aura’ of neurological symptoms localizable to the cerebral cortex or brainstem, such as visual disturbance, paraesthesia, diplopia or limb weakness. These develop gradually over 5–20 minutes and last less than 60 minutes. Headache, nausea and/or photophobia usually follow after an interval of less than an hour. Several variant forms of migraine have been defined, including ophthalmoplegic, abdominal, hemiplegic and retinal migraine, but all are uncommon. In ophthalmoplegic migraine, the headache is associated with paralysis of one or more of the nerves supplying the ocular muscles. Horner’s syndrome may also occur. Abdominal migraine manifests as recurrent episodes of abdominal pain for which no other cause is found. Retinal migraine, which is fortunately very rare, involves recurrent attacks of retinal ischaemia which may lead to bilateral optic atrophy. Hemiplegic migraine is a stroke mimic. The complexity of the mechanisms involved in the genesis of migraine suggests that there are a number of ways to interrupt the processes to provide effective relief from symptoms. A wide variety of pharmacological agents and combinations of agents have been tried for the treatment of migraine, with varying results. Interpreting the evidence is challenging, as the majority of the studies have small sample sizes, compare different agents or combinations of agents, are conducted in settings other than EDs and the outcome measure(s) tested varies widely. For mild to moderate migraine headache in patients who have not taken other medication, aspirin 900 mg combined with metoclopramide 10 mg is effective. Most ED patients, however, have either tried their usual medication or have significant nausea or vomiting making oral therapy inappropriate. The effectiveness of commonly used agents is summarized in Table 8.1.3. Dosing and administration are summarized in Table 8.1.4. At present, the most effective agents appear to be the phenothiazines (chlorpromazine and prochlorperazine) and the triptans, each of which has achieved>70% efficacy in a number of studies. Note that triptans are contraindicated in patients with a history of ischaemic heart disease, uncontrolled hypertension or with the concomitant use of ergot preparations. Table 8.1.3 Pooled effectiveness data from ED studies of the treatment of migraine Table 8.1.4 Drug dosing and administration Pethidine (meperidine) is not indicated for the treatment of migraine. Its reported effectiveness is only 56%, it has a high rate of rebound headache and it carries a risk of dependence. The data on dihydroergotamine are difficult to interpret because it is often used in combination with other agents (e.g. metoclopramide); however, it has also been shown to be less effective than chlorpromazine and sumatriptan in acute treatment and to have a high rate of unpleasant side effects. Sodium valproate and haloperidol have also shown moderate effectiveness in small studies, but there are insufficient data to draw a valid conclusion or recommend them as treatment options. Lignocaine (lidocaine) has been shown to be no more effective than placebo. The efficacy of intravenous magnesium sulphate (1 or 2 mg) remains unclear. It was shown in a small placebo-controlled trial to be effective but, in another study, the combination of magnesium with metoclopramide was less effective than metoclopramide and placebo. Rebound or recurrent headache is common in ED patients treated for migraine (approximately 30%). There is evidence that oral or IV dexamethasone, in addition to standard migraine therapy for selected patients, reduces the proportion of patients who experience early recurrence (so-called rebound headache). A meta-analysis of published papers reports a 26% reduction in the relative risk of headache recurrence within 72 hours. Doses used were 10 mg IV or 8 mg orally. Trigeminal neuralgia is a debilitating condition in which patients describe ‘lightning-’or a ‘hot poker-’like pain that is severe and follows the distribution of the trigeminal nerve. Individual episodes of pain last only seconds, but may recur repeatedly within a short period and can be triggered by minor stimuli, such as light touch, eating or drinking, shaving or passing gusts of wind. It is most common in middle or older age. Evidence suggests that the pathological basis of trigeminal neuralgia is demyelination of sensory fibres of the trigeminal nerve in the proximal (CNS) portion of the nerve root or, rarely, in the brainstem, most commonly due to compression of the nerve root by an overlying artery or vein. Trigeminal neuralgia is classified as classic trigeminal neuralgia (no cause identified) and symptomatic trigeminal neuralgia (secondary to another condition). Characteristics associated with symptomatic trigeminal neuralgia are trigeminal sensory deficits and bilateral involvement. In approximately 15% of cases, there is a structural cause for trigeminal neuralgia. For this reason, there is some support for routine neuroimaging (CT, MRI) in these patients. Electrophysiological assessment of trigeminal reflexes can also be helpful in distinguishing classic from symptomatic trigeminal neuralgia. The choice between the two approaches will depend on availability, expertise, cost and patient and treating clinician preference. The mainstay of therapy for trigeminal neuralgia is carbamazepine. The usual starting dose is 200–400 mg/day in divided doses, increased by 200 mg/day until relief up to a maximum of 1200 mg/day. The average dose required is 800 mg/day. Where available, oxcarbazepine 600–1800 mg/day is an effective alternative. For patients who fail first-line therapy, there is some evidence to support the addition of lamotrigine or a change to baclofen. Referral for consideration of surgery is appropriate in patients who are refractory to medical therapy. Giant cell arteritis is the most common form of vasculitis in patients aged over 50 years. It affects large and middle-sided blood vessels with a predisposition for the cranial arteries arising from the carotid arteries. Loss of vision is the most common severe complication. Involvement of extracranial arteries including the aorta is more frequent than previously assumed. Inflammation markers in blood are usually elevated, but specific laboratory tests for the diagnosis of giant cell arteritis are not available. Imaging using ultrasonography, magnetic resonance imaging and positron emission tomography can be useful to confirm, localize and assess the extent of vascular involvement. Temporal artery biopsy is the gold standard for diagnosis. Glucocorticoids are still the standard therapy (50–100 mg/day). Patients with acute visual changes secondary to giant cell arteritis should receive parenteral corticosteroid therapy and be admitted until their condition stabilizes. Controversies Philip Aplin and Mark Morphett Cerebrovascular disease is the third highest cause of death in developed countries, after heart disease and cancer. A stroke is an acute neurological injury secondary to cerebrovascular disease, either by infarction (80%) or by haemorrhage (20%). The incidence of stroke is steady and, although mortality is decreasing, it is still a leading cause of long-term disability. Transient ischaemic attacks (TIAs) are defined as transient episodes of neurological dysfunction caused by focal brain, spinal cord or retinal ischaemia, without acute infarction. Causes are similar to those of ischaemic stroke, particularly atherosclerotic thromboembolism related to the cerebral circulation and cardioembolism. Diagnosis of the cause of TIAs with appropriate management is important in order to prevent a potentially devastating stroke. Brain tissue is very sensitive to the effects of oxygen deprivation. Following cerebral vascular occlusion, a series of metabolic consequences may ensue, depending on the extent, duration and vessels involved, which can lead to cell death. Reperfusion of occluded vessels may also occur, either spontaneously or via therapeutic intervention, with the potential for reperfusion injury. An area of threatened but possibly salvageable brain may surround an area of infarction. The identification of this so-called ischaemic penumbra and therapeutic efforts to ameliorate the extent of irreversible neuronal damage, have been the subject of ongoing research efforts. Large anterior circulation ischaemic strokes can be associated with increasing mass effect and intracranial pressure in the hours to days following onset. Secondary haemorrhage into an infarct may also occur, either spontaneously or related to therapy. Clinical deterioration often follows. These are the results of several pathological processes (Table 8.2.1): Table 8.2.1 Ischaemic stroke Arterial thromboembolism Carotid and vertebral artery atheroma Intracranial vessel atheroma Small vessel disease – lacunar infarction Haematological disorders – hypercoagulable states Cardioembolism Aortic and mitral valve disease Atrial fibrillation Mural thrombus Atrial myxoma Paradoxical emboli Hypoperfusion Severe vascular stenosis or a combination of these factors Hypotension Vasoconstriction – drug induced, post-SAH, pre-eclampsia Other vascular disorders Arterial dissection Gas embolism syndromes Moyamoya disease Arteritis Intracerebral haemorrhage Hypertensive vascular disease Lipohyalinosis and microaneurysms Aneurysms Saccular Mycotic Arteriovenous malformations Amyloid angiopathy Bleeding diathesis Anticoagulation Thrombolytics Thrombocytopenia/disseminated intravascular coagulation Haemophilia Secondary haemorrhage into a lesion – tumour or infarction Haemorrhagic stroke is the result of vessel rupture into the surrounding intracerebral tissue or subarachnoid space. Subarachnoid haemorrhage is the subject of a separate chapter in this book (see Chapter 8.3). The neurological defect associated with an intracerebral haemorrhage (ICH) is the consequence of direct brain injury, secondary occlusion of nearby vessels, reduced cerebral perfusion caused by associated raised intracranial pressure and cerebral herniation. The causes of ICH include: This particularly applies to cerebral ischaemic events, both TIAs and strokes. Non-modifiable risk factors for ischaemic stroke include: In terms of primary prevention, hypertension is the most important modifiable risk factor. The benefit of antihypertensive treatment in stroke prevention has been well shown. The other major risk factors for atherosclerosis and its complications – diabetes, smoking and hypercholesterolaemia – often contribute to increased stroke risk. These should be managed according to standard guidelines. The most important cardiac risk factor for TIA and stroke is atrial fibrillation (AF), both chronic and paroxysmal. Warfarin is recommended to prevent cardioembolism where the risk:benefit ratio of anticoagulation (target INR 2.0–3.0) favours this. Prediction tools, such as the CHADS2 and CHA2DS2-VASc scores, have been developed to standardize the approach to primary stroke prevention in patients with non-valvular AF. Recently, an oral direct thrombin inhibitor (dabigatran) has been shown to be non-inferior to warfarin for stroke prevention in a large industry sponsored trial (the RE-LY trial). On the basis of this trial, dabigatran has been approved for use as an alternative to warfarin with rapid uptake of this medication in the community. Those with contraindications to warfarin or very low stroke risk should initially receive aspirin. A carotid bruit or carotid stenosis found in an otherwise asymptomatic patient is associated with an increased stroke risk. However, the role of carotid endarterectomy in these patients is controversial. While early trials suggested some minor benefit, more recent studies have refuted this and it is increasingly clear that intensive medical therapy in patients with asymptomatic carotid stenosis reduces stroke risk well below that achieved with either endarterectomy or carotid stenting. Other major cardiac conditions associated with increased TIA/stroke risk include endocarditis, mitral stenosis, prosthetic heart valves, recent myocardial infarction and left ventricular aneurysm. Less common ones include atrial myxoma, a patent foramen ovale and cardiomyopathies. Secondary prevention involves detection and modification, if possible, of conditions that may have caused a TIA or stroke in order to prevent further events that may result in worse clinical outcomes. As well as the risk factors already mentioned, many other uncommon conditions, such as arterial dissection and prothrombotic states, may cause TIA and stroke. These will be discussed later in the chapter. The symptoms and signs of stroke or TIA correspond to the area of the brain affected by ischaemia or haemorrhage (Table 8.2.2). Table 8.2.2 *Usually regarded as carotid distribution. In ischaemic brain injury, the history and pattern of physical signs may correspond to a characteristic clinical syndrome according to the underlying cause and the vessel occluded. This has a bearing on the direction of further investigation and treatment decisions. Differentiating between anterior and posterior circulation ischaemia/infarction is important in this respect, but is not always possible on clinical grounds alone. Determining the cause of the event is the next step. Once again, clues, such as a carotid bruit or atrial fibrillation, may be present on clinical evaluation. For accurate delineation of the site of the brain lesion, exclusion of haemorrhage and assessment of the underlying cause, it is usually necessary to undertake imaging studies. The anterior circulation supplies blood to 80% of the brain and consists of the ICA and its branches, principally the ophthalmic, middle cerebral and anterior cerebral arteries. This system supplies the optic nerve, retina, frontoparietal and most of the temporal lobes. Ischaemic injury involving the anterior cerebral circulation commonly has its origins in atherothrombotic disease of the ICA. Atherosclerosis of this artery usually affects the proximal 2 cm, just distal to the division of the common carotid artery. Advanced lesions may be the source of embolism to other parts of the anterior circulation or cause severe stenosis with resultant hypoperfusion distally if there is inadequate collateral supply via the circle of Willis. This is usually manifest by signs and symptoms in the middle cerebral artery (MCA) territory (Table 8.2.3). Less commonly, lesions of the intracranial ICA and MCA may cause similar clinical features. Table 8.2.3 Signs of middle cerebral artery (MCA) occlusion Homonymous hemianopia Contralateral hemiplegia affecting face and arm more than leg Contralateral hemisensory loss Dysphasias with dominant hemispheric involvement (usually left) Spatial neglect and dressing apraxia with non-dominant hemispheric involvement Embolism to the ophthalmic artery or its branches causes monocular visual symptoms of blurring, loss of vision and field defects. When transient, this is referred to as amaurosis fugax or transient monocular blindness. The anterior cerebral artery territory is the least commonly affected by ischaemia because of the collateral supply via the anterior communicating artery. If occlusion occurs distally or the collateral supply is inadequate, then ischaemia may occur. This manifests as sensory/motor changes in the leg – more so than in the arm. More subtle changes of personality may occur with frontal lobe lesions, as may disturbances of micturition and conjugate gaze. Major alterations of consciousness, with Glasgow coma scores<8, imply bilateral hemispheric or brainstem dysfunction. The brainstem may be primarily involved by a brainstem stroke or secondarily affected by an ischaemic or haemorrhagic lesion elsewhere in the brain, owing to a mass effect and/or increased intracranial pressure. Ischaemic injury in the posterior circulation involves the vertebrobasilar arteries and their major branches which supply the cerebellum, brainstem, thalamus, medial temporal and occipital lobes. Posterior cerebral artery occlusion is manifested by visual changes of homonymous hemianopia (typically with macular sparing if the MCA supplies this part of the occipital cortex). Cortical blindness, of which the patient may be unaware, occurs with bilateral posterior cerebral artery infarction. Depending on the area and extent of involvement, brainstem and cerebellar stroke manifest as a combination of: motor and sensory abnormalities, which may be uni- or bilateral; cerebellar features of vertigo, nystagmus and ataxia; and cranial nerve signs, such as diplopia/ophthalmoplegia, facial weakness and dysarthria. Consciousness may also be affected. Examples of brainstem stroke patterns include (this list is by no means exhaustive): Lacunar infarcts are associated primarily with hypertension and diabetes. They occur in the small penetrating arteries supplying the internal capsule, thalamus and upper brainstem. Isolated motor or sensory deficits are most commonly seen. This includes the circumstances, time of onset, associated symptoms, such as headache, and any resolution/progression of signs and symptoms. It may be necessary to take a history from a relative or friend, particularly in the presence of dysphasia or reduced conscious state. The history of a stroke is usually of acute onset of a neurological deficit over minutes but, occasionally, there may be a more gradual or stuttering nature to a presentation over a period of hours. A past history of similar events suggestive of a TIA should be carefully sought. The presence of a severe headache with the onset of symptoms may indicate ICH or SAH. However, headache may also occur with ischaemic strokes. A declining level of consciousness may indicate increasing intracranial pressure due to an ICH or a large anterior circulation infarct – so-called malignant MCA infarction. It may also be caused by pressure on the brainstem by an infratentorial lesion, such as a cerebellar haemorrhage. The possibility of trauma or drug abuse should be remembered along with the past medical and medication history, particularly anticoagulant/antiplatelet therapy. Risk factors for vascular disease, cardiac embolism, venous embolism and increased bleeding should be sought. In young patients with an acute neurological deficit, dissection of the carotid or vertebral artery should be considered. This is often associated with neck pain and headaches/facial pain with or without a history of neck trauma. Trauma if present may be minor, such as a twisting or hyperextension/flexion injury sustained in a motor vehicle accident, playing sports or neck manipulation. Cardioembolism tends to produce ischaemic injury in different parts of the brain, resulting in non-stereotypical recurrent TIAs, whereas atherothrombotic disease of the cerebral vessels tends to cause recurrent TIAs of a similar nature, particularly in stenosing lesions of the internal carotid or vertebrobasilar arteries. This includes assessing the level of consciousness, pupil size and reactivity, extent of neurological deficit, presence of neck stiffness and funduscopy for signs of papilloedema and retinal haemorrhage. Quantifying the neurological deficit using a stroke scale, such as the 42-point National Institute of Health Stroke Scale (NIHSS), is useful in the initial assessment and also for monitoring progress in a more objective way than clinical description alone. Strokes with a NIHSS score>22 are classified as severe. In the case of TIA, all clinical signs may have resolved. The average TIA lasts less than 15 minutes. This includes carotid auscultation and is directed towards findings associated with a cardioembolic source. A carotid bruit in a symptomatic patient is likely to predict a moderate to severe carotid stenosis. Conversely, the absence of a carotid bruit does not exclude significant carotid artery disease as a cause of a TIA or stroke. Major risk factors for cardioembolism that can be identified in the emergency department (ED) include AF, mitral stenosis, prosthetic heart valves, infective endocarditis, recent myocardial infarction, left ventricular aneurysm and cardiomyopathies. The acute onset of stroke and TIA is characteristic, however, misdiagnoses (the so-called ‘stroke mimics’) can occur. The most common stroke mimics are seizures (particularly when there is associated Todd’s paresis), hypoglycaemia, systemic infection, brain tumour and toxic metabolic disorders. Others include subdural haematoma, hypertensive encephalopathy, encephalitis, multiple sclerosis, migraine and conversion disorder. This has implications when considering more aggressive stroke interventions, such as thrombolysis. Table 8.2.4 Differential diagnosis of stroke Intracranial space-occupying lesion Subdural haematoma Brain tumour Brain abscess Postictal neurological deficit – Todd’s paresis Head injury Encephalitis Metabolic or drug-induced encephalopathy Hypoglycaemia, hyponatraemia, etc. Wernicke–Korsakoff syndrome Drug toxicity Hypertensive encephalopathy Multiple sclerosis Migraine Peripheral nerve lesions Functional
Neurology Emergencies
8.1 Headache
Introduction
Aetiology, pathophysiology and pathology
 the great venous sinuses and their branches and
the great venous sinuses and their branches and
 the basal dura and dural arteries, but to a lesser extent than the other structures.
the basal dura and dural arteries, but to a lesser extent than the other structures.
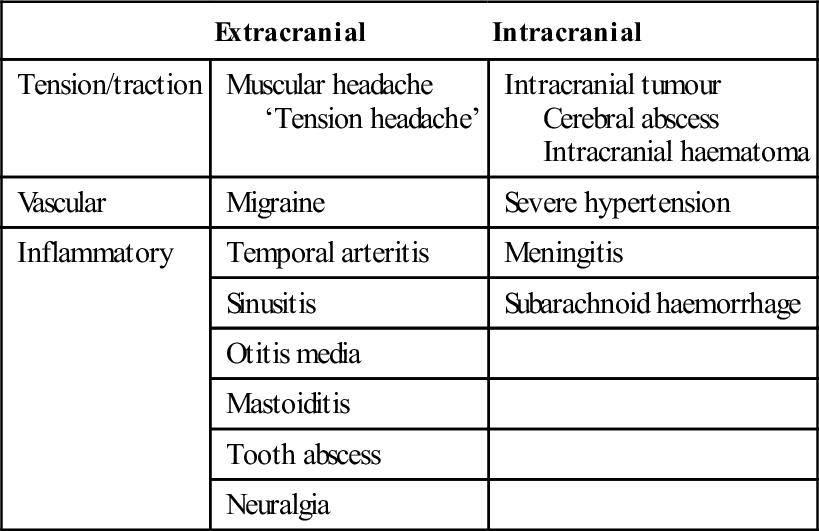
Extracranial
Intracranial
Tension/traction
Muscular headache
‘Tension headache’
Intracranial tumour
Cerebral abscess
Intracranial haematoma
Vascular
Migraine
Severe hypertension
Inflammatory
Temporal arteritis
Meningitis
Sinusitis
Subarachnoid haemorrhage
Otitis media
Mastoiditis
Tooth abscess
Neuralgia
Clinical features
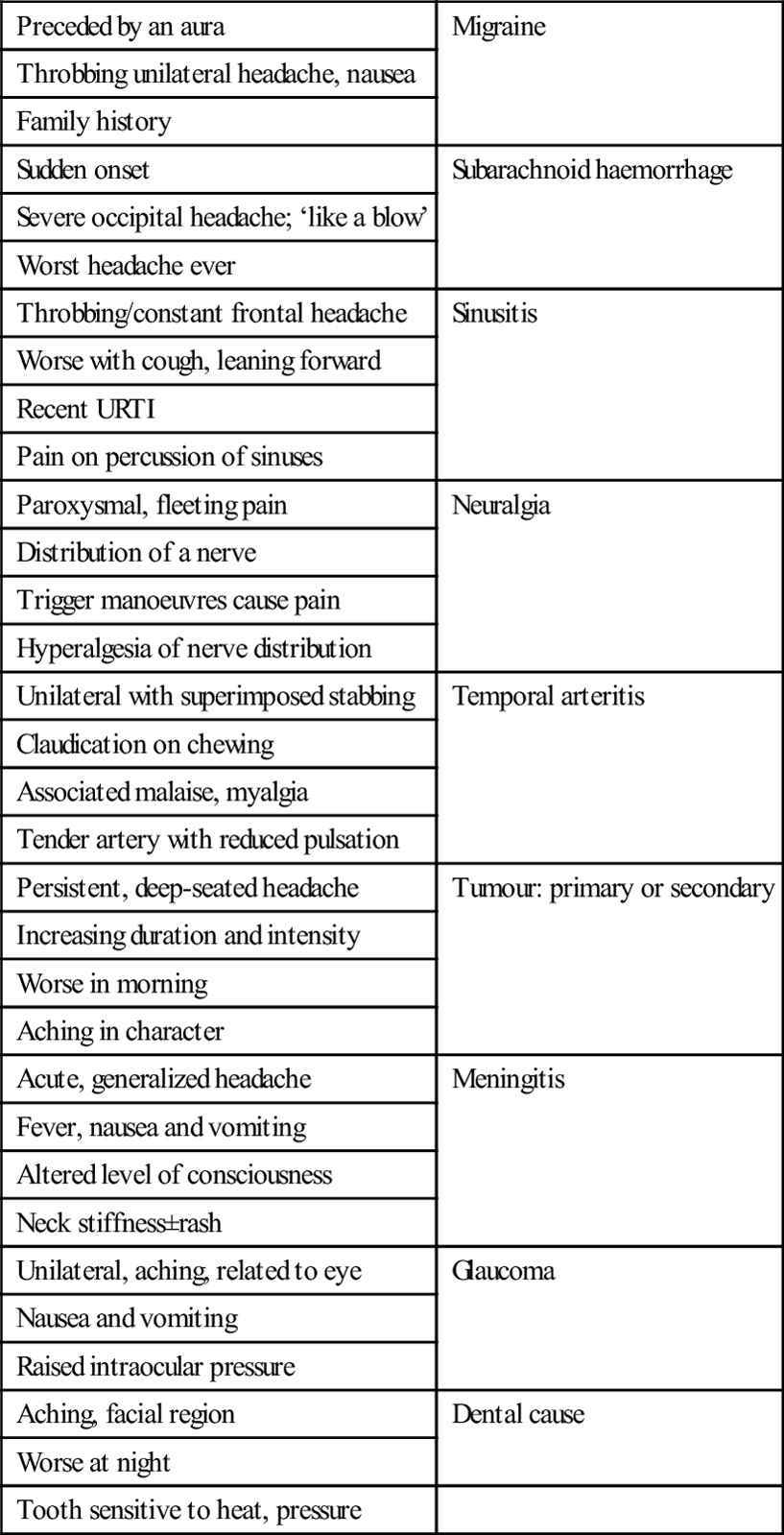
Headache patterns
Preceded by an aura
Migraine
Throbbing unilateral headache, nausea
Family history
Sudden onset
Subarachnoid haemorrhage
Severe occipital headache; ‘like a blow’
Worst headache ever
Throbbing/constant frontal headache
Sinusitis
Worse with cough, leaning forward
Recent URTI
Pain on percussion of sinuses
Paroxysmal, fleeting pain
Neuralgia
Distribution of a nerve
Trigger manoeuvres cause pain
Hyperalgesia of nerve distribution
Unilateral with superimposed stabbing
Temporal arteritis
Claudication on chewing
Associated malaise, myalgia
Tender artery with reduced pulsation
Persistent, deep-seated headache
Tumour: primary or secondary
Increasing duration and intensity
Worse in morning
Aching in character
Acute, generalized headache
Meningitis
Fever, nausea and vomiting
Altered level of consciousness
Neck stiffness±rash
Unilateral, aching, related to eye
Glaucoma
Nausea and vomiting
Raised intraocular pressure
Aching, facial region
Dental cause
Worse at night
Tooth sensitive to heat, pressure
Clinical investigations
Tension headache
Migraine
Pathophysiology
Classification and clinical features
Treatment
Agent
No. of studies
Total patients
Clinical success rate (%)
NNT: clinical success
Chlorpromazine (IV)
6
171
85
1.67
Prochlorperazine (IM or IV)
6
171
71
2.17
Sumatriptan (6 mg SC)
21
3139
71
2.17
Ketorolac (IM or IV)
6
155
66
2.44
Tramadol (IM or IV)
3
191
60
2.86
Metoclopramide (IV)
7
300
58
3.03
Magnesium sulphate (IV)
3
117
41
6.25
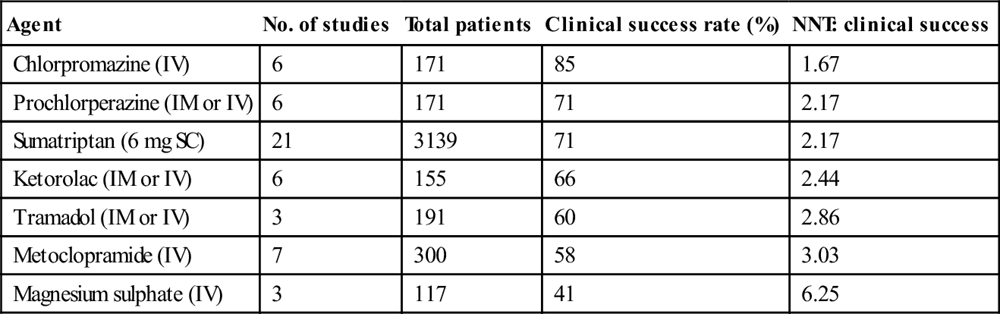
Agent
Drug dosing/administration
Chlorpromazine (IV)
12.5 mg intravenously, repeated every 20 minutes as needed to a maximum dose of 37.5 mg, accompanied by 1 L normal saline over 1 hour to avoid hypotension
OR
25 mg in 1 L normal saline over 1 hour, repeated if necessary
Prochlorperazine (IM or IV)
10 mg/12.5 mg (depending on packaging)
Sumatriptan (SC, IN)
6 mg SC, 20 mg IN
Metoclopramide (IV)
10–20 mg
Ketorolac (IM or IV)
30 mg IV, 60 mg IM
Tramadol (IM)
100 mg
Trigeminal neuralgia
Aetiology and pathophysiology
Clinical investigations
Treatment
Temporal (giant cell) arteritis
8.2 Stroke and transient ischaemic attacks
Introduction
Pathophysiology
Ischaemic strokes
Haemorrhagic stroke
 Haemorrhage into an underlying lesion, e.g. tumour or infarction.
Haemorrhage into an underlying lesion, e.g. tumour or infarction.
 Drug toxicity from sympathomimetics and cocaine.
Drug toxicity from sympathomimetics and cocaine.
Risk factors for TIA/stroke and prevention
 increasing age: the stroke rate more than doubles for each 10 years above age 55.
increasing age: the stroke rate more than doubles for each 10 years above age 55.
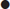 gender: stroke is slightly more common in males than females.
gender: stroke is slightly more common in males than females.
Ischaemic stroke syndromes
Arterial territory
Symptom
Carotid
Either
Vertebrobasilar
Dysphasia
+
Monocular visual loss
+
Unilateral weakness*
+
Unilateral sensory disturbance*
+
Dysarthria**
+
Homonymous hemianopia
+
Dysphagia**
+
Diplopia**
+
Vertigo**
+
Bilateral simultaneous visual loss
+
Bilateral simultaneous weakness
+
Bilateral simultaneous sensory disturbance
+
Crossed sensory/motor loss
+
Patterns of clinical features
Anterior circulation ischaemia
Posterior circulation ischaemia
Lacunar infarcts
Clinical features
History
Examination
Central nervous system
Cardiovascular
Differential diagnosis (Table 8.2.4)
Related posts:

Full access? Get Clinical Tree



8. Neurology Emergencies
Only gold members can continue reading. Log In or Register to continue