Simple and inexpensive interventions in the ICU, such as the utilization of checklists and strict attention to aseptic technique during central venous catheterization, can result in substantial improvements in patient outcomes.
 Administration of high-dose corticosteroids to patients presenting with traumatic brain injury is associated with 20% increase in the relative risk of death.
Administration of high-dose corticosteroids to patients presenting with traumatic brain injury is associated with 20% increase in the relative risk of death.
 Administration of thrombolytic therapy (rt-PA) to patients presenting within 4.5 hours of onset of acute ischemic stroke results in improved neurologic outcome.
Administration of thrombolytic therapy (rt-PA) to patients presenting within 4.5 hours of onset of acute ischemic stroke results in improved neurologic outcome.
 Patients who are resuscitated from cardiac arrest due to ventricular fibrillation have improved neurologic outcome and possibly reduced mortality when treated with mild therapeutic hypothermia (32° to 34°C) for 12 to 24 hours after hospital admission.
Patients who are resuscitated from cardiac arrest due to ventricular fibrillation have improved neurologic outcome and possibly reduced mortality when treated with mild therapeutic hypothermia (32° to 34°C) for 12 to 24 hours after hospital admission.
 In patients with severe sepsis or septic shock, early and aggressive use of fluid resuscitation, appropriate antibiotics, infectious source control, and vasopressors/inotropes as needed improves survival.
In patients with severe sepsis or septic shock, early and aggressive use of fluid resuscitation, appropriate antibiotics, infectious source control, and vasopressors/inotropes as needed improves survival.
 Separation from mechanical ventilation in patients who are recovering from respiratory failure is accelerated by respiratory therapy–driven protocols and daily trials of spontaneous breathing.
Separation from mechanical ventilation in patients who are recovering from respiratory failure is accelerated by respiratory therapy–driven protocols and daily trials of spontaneous breathing.
 Ventilation with low tidal volume (6 mL/kg) in patients with acute lung injury and acute respiratory distress syndrome reduces mortality, compared to traditional tidal volumes (12 mL/kg).
Ventilation with low tidal volume (6 mL/kg) in patients with acute lung injury and acute respiratory distress syndrome reduces mortality, compared to traditional tidal volumes (12 mL/kg).
 Red blood cell transfusion in the ICU should be restricted (transfusion threshold hemoglobin <7 g/dL) with the possible exception of patients with a diagnosis of active bleeding, early septic shock, acute myocardial infarction, or unstable angina, or with primarily neurologic or neurosurgical problems.
Red blood cell transfusion in the ICU should be restricted (transfusion threshold hemoglobin <7 g/dL) with the possible exception of patients with a diagnosis of active bleeding, early septic shock, acute myocardial infarction, or unstable angina, or with primarily neurologic or neurosurgical problems.
 Nurse-driven sedation protocols and reduction of sedation reduce the duration of mechanical ventilation and ICU length of stay.
Nurse-driven sedation protocols and reduction of sedation reduce the duration of mechanical ventilation and ICU length of stay.
 The incidence of ventilator-associated pneumonia (VAP) can be reduced with strict hand washing during patient care and semirecumbent positioning of the patient. Antibiotic therapy of VAP should utilize a “de-escalating” strategy, and can be limited to an 8-day course in uncomplicated cases.
The incidence of ventilator-associated pneumonia (VAP) can be reduced with strict hand washing during patient care and semirecumbent positioning of the patient. Antibiotic therapy of VAP should utilize a “de-escalating” strategy, and can be limited to an 8-day course in uncomplicated cases.
Multimedia
 Cerebral Blood Flow
Cerebral Blood Flow
 Swan Pressure Tracing
Swan Pressure Tracing
 Swan Real Life
Swan Real Life
 A-line Insertion
A-line Insertion
 Mechanical Ventilation Pressure
Mechanical Ventilation Pressure
 A-line Infection
A-line Infection
INTRODUCTION: ANESTHESIOLOGISTS AND CRITICAL CARE MEDICINE
Historically, Critical Care Medicine evolved as a specialty nearly simultaneously in Europe and North America, but has followed strikingly different models in regards to the involvement of anesthesiologists. The first intensive care unit (ICU) in Europe may have been located in Denmark in the 1950s, and concurrently the first critical care physician, or “intensivist,” may well have been an anesthesiologist.1 Anesthesiologists continued to play a defining role in the development of Critical Care Medicine in most of Europe, Australia, New Zealand, Japan, and elsewhere and comprise the majority of intensivists in many countries around the world today. In North America, anesthesiologists were also integral to the development of Critical Care Medicine as a specialty. However, in contrast to other countries, in the United States anesthesiologists have played an ever-diminishing role in the specialty, and today comprise a small minority of the intensivist workforce.2
Although it has been suggested that the first ICU in North America was established at Johns Hopkins in 1923 to care for postoperative neurosurgical patients, it was not until the late 1950s and early 1960s that true multidisciplinary ICUs began to appear. The driving forces behind ICU development included advances in surgical techniques, polio epidemics which resulted in widespread respiratory failure, and later the recognition of the acute respiratory distress syndrome (ARDS). Anesthesiologists played a natural role in the evolution of ICUs, given their familiarity with surgical resuscitation and mechanical ventilation. Early on, however, the concept of “intensivists” did not exist, and patients were often managed by their primary physician (be it a surgeon or an internist) and nurses, with formal or informal consultation given by specialists, including anesthesiologists.
In the early 1960s, the first Critical Care Medicine training program was established at the University of Pittsburgh under the direction of an anesthesiologist, Peter Safar. At this point, the concept of “intensivist” was born; as defined by Dr. Safar, the qualities and qualifications of such an individual should include inquisitiveness, thoughtfulness, a high level of motivation, action orientation, diplomacy, and scientific training. In the late 1960s, a group including Dr. Safar and another anesthesiologist, Ake Grenvik, were instrumental in inaugurating the Society of Critical Care Medicine (SCCM). Anesthesiologists working through SCCM were instrumental in developing the board certification process for Critical Care Medicine, and in 1986 the first Critical Care Medicine Certification examination was administered by the American Board of Anesthesiology.3 From the 1960s until the present, numerous anesthesiologists made important contributions to the development of the specialty, to critical care–related research, and to improvements in the care of critically ill patients. However, as of 2010 only about 1,300 anesthesiologists had completed the certification process in Critical Care Medicine in the United States, and anesthesiologists comprised only 8% of adult intensivists certified in the past 10 years.4
Anesthesiology and Critical Care Medicine: The Future
Although it seems certain that anesthesiologists will continue to play an important role in Critical Care Medicine worldwide, in the United States anesthesiology is currently at a crossroads in regards to its continued involvement in Critical Care Medicine. There are several forces that will shape the evolution of the specialty of Critical Care Medicine as a whole and the contribution that anesthesiologists will make to this evolution: (1) Increasing evidence that intensivists increase the quality of care and improve outcomes; (2) business/economic factors, whereby employers, insurers, and hospitals recognize the cost savings associated with intensivists in the ICU, as exemplified by the Leapfrog Initiative (further discussed in the following section); and (3) the aging population and increasing demand for critical care services, which will result in a shortage of intensivists starting in 2007 that will grow to a greater than 20% deficit by the year 2020, given current training levels2 (Table 55-1). This prediction does not take into account any additional demands placed on the healthcare system, if any, by the Leapfrog Initiative and thus is likely an underestimate of the true future need for intensivists.
Given the above observations, it is clear that opportunities for careers in Critical Care Medicine will be amply available in the coming years. Anesthesiology as a specialty would seem ideally suited to help satisfy the increasing demand for intensivists. Anesthesiologists are hospital based; have sound fundamental training in physiology, pharmacology, invasive procedures, and monitoring; and have excellent historical and concurrent role models for the anesthesiologist as intensivist. However, economic and lifestyle incentives have recently dissuaded anesthesiology trainees from pursuing further training and careers in Critical Care Medicine. Critical care services are currently not reimbursed at rates commensurate with surgical anesthesia, and critical care practice is accurately perceived as more time-consuming and the workload more unpredictable than an operating room–based practice. However, these factors may change in the coming years, as reimbursement for surgical anesthesia and critical care services equalize and the lifestyle constraints associated with critical care are moderated by creative organizational strategies, including the use of physician extenders and other mechanisms for reducing the “24–7” workload. Lastly, the increase in the anesthesiology residency training requirement to include 4 months of intensive care may imbue trainees with a greater interest in Critical Care Medicine as a career choice. As stated above, anesthesiology in the United States is at a crossroads; with proper imagination, emphasis, support, and training the specialty can reassert itself as a leader in the field of Critical Care Medicine.
TABLE 55-1. LEAPFROG ICU PHYSICIAN STAFFING (IPS) STANDARD

Critical Care Medicine: A Systems and Evidence-based Approach
Critical care encompasses all disciplines of medicine. It is clearly beyond the scope of a single chapter to provide detailed coverage of all aspects of critical illness, including physiology, pathophysiology, and management of disease. In addition, many critical care issues are commonly encountered by anesthesiologists that practice solely in the operating room and are covered in detail elsewhere in this text. Thus, this chapter will focus on topics that are relatively unique to the ICU, on therapeutic approaches, and upon practices for which strong evidence exists, particularly when supported by randomized controlled trials (RCTs).
Grading of levels of evidence and practice guidelines in an effort to improve clinician confidence in recommendations has become standard practice. Several different grading systems exist, with no clear evidence that one is superior to another; furthermore, given uncertainty about the methodology of grading systems and their effects on patient outcomes,5 we have chosen not to include “grades” or levels of evidence in this chapter.
PROCESS OF CARE IN THE ICU
The method by which care is delivered may affect outcomes as much or more than the specific interventions employed. For example, care delivery models that reduce errors and encourage utilization of evidence-based practices may have a profound effect on outcomes, in addition to reducing costs. A single adverse event in the ICU may cost as much as $4,000 and increase length of stay by 1 day.6 Additional data suggest that implementation of evidence-based practices in the ICU is not consistent and that more uniform implementation could save up to approximately 200,000 lives per year in the United States.7
As advances in medical and surgical therapeutics have increased the complexity of care for an aging and increasingly ill population of patients, it has become increasingly clear that the involvement of intensivists in the management of the critically ill is desirable. Several studies have suggested that mortality and other intermediate end points such as ICU length of stay can be reduced when “high-intensity” physician staffing models that mandate management or comanagement by intensivists are used.8 These studies, as summarized in a meta-analysis by Pronovost et al. and further supported by two subsequent cohort studies,8–10 have provoked a reconsideration of the ideal staffing model for ICUs in the United States, particularly amongst the business community.
The Business Roundtable, a national association of CEOs of Fortune 500 companies, formed the Leapfrog Group in 1999. The Leapfrog Group is a coalition of over 150 purchasers and providers of healthcare benefits, including large companies such as General Motors, Motorola, and Merck and insurers such as Aetna. The stated goal of the Leapfrog Group is to improve health care, in particular by reducing deaths due to medical error. To accomplish this mission, the group formulated the Leapfrog Initiative, which includes a series of “safety standards” that healthcare providers (largely hospitals) should strive for if they are to provide care for Leapfrog Group employees. Prompted by the data associating intensivists with improved outcomes, the Leapfrog Initiative included an ICU Physician Staffing (IPS) standard that promotes the continuous involvement of intensivists in the care of critically ill patients (Table 55-1).11 Less than 10% of hospitals met this standard prior to its publication, and a recent survey suggests that there has been little improvement in compliance since then.12,13 Reported barriers to implementation of the Leapfrog Initiative include lack of ICU directors, cost, loss of control and continuity in patient care, and loss of income.13 However, considerable cost savings as a result of Leapfrog Initiative implementation have been estimated which continues to provide additional impetus for widespread adoption of these practices.14
 In addition to improved ICU staffing, other simple interventions in the process of care have been shown to improve outcomes. These include efforts to reduce errors of omission by utilizing checklists during procedures and patient rounds,15 and by explicitly outlining the daily goals of care during rounds.16 Such efforts incur little or no cost, may incur substantial benefits, but are severely underutilized in the United States at present.
In addition to improved ICU staffing, other simple interventions in the process of care have been shown to improve outcomes. These include efforts to reduce errors of omission by utilizing checklists during procedures and patient rounds,15 and by explicitly outlining the daily goals of care during rounds.16 Such efforts incur little or no cost, may incur substantial benefits, but are severely underutilized in the United States at present.
NEUROLOGIC AND NEUROSURGICAL CRITICAL CARE
Neuromonitoring
Several neuromonitoring devices used in the ICU setting may help in assessing pathophysiologic processes and adjusting therapy. The following section will discuss some commonly used neuromonitoring devices, including transcranial Doppler (TCD) ultrasonography, brain tissue oxygenation (PbrO2), and microdialysis. Intracranial pressure (ICP) monitoring and jugular venous oximetry are discussed in chapters on Standard Monitoring Techniques, Anesthesia for Neurosurgery, and Burns and Trauma, and will not be discussed in detail here.
Transcranial Doppler Ultrasonography
TCD measures blood flow velocities and can indirectly estimate cerebral blood flow, ICP, cerebral autoregulation, and intracranial compliance. In patients with subarachnoid hemorrhage (SAH) or traumatic brain injury (TBI) TCD can be used as a tool to identify vasospasm as flow velocities through a given blood vessel tend to increase during spasm. In patients with TBI, flow velocities are depressed, and impaired autoregulation and vascular reactivity are common. In these patients, monitoring of TCD and jugular venous oxygen saturation (SjO2) (see later) may be used to define the optimum cerebral perfusion pressure (CPP).17 In addition, TCD indices may predict neurologic outcome in patients with TBI.18 While anatomic variations preclude obtaining TCDs in all patients, the major barrier to more widespread adoption of this modality is the expertise required to obtain and interpret the data accurately.
Brain Tissue Oxygenation
Brain tissue oxygen pressure (PbrO2) measurements are performed by introducing a small, oxygen-sensitive catheter into the brain tissue. The device monitors a very local area of the brain tissue, and this technique is increasingly used for evaluation of cerebral oxygenation (normal PbrO2 values: 25 to 30 mm Hg).19 Monitoring may be performed in relatively undamaged parts of the brain or, preferably, in the penumbra region of an intracerebral lesion.20 Various studies have shown that an increase in ICP and a decrease in CPP or arterial oxygenation, and hyperventilation may result in decreased PbrO2. This monitoring modality has been shown to be helpful in detecting intraoperative brain hypoxia during episodes of hyperventilation.21 In patients with TBI, ischemic episodes defined as PbrO2 <10 mm Hg for longer than 15 minutes in the first week after the injury were found to be associated with unfavorable neurologic outcome, and values of 0 (not responding to an oxygen challenge) are compatible with brain death.22 A PbrO2 of 15 mm Hg or less has been recommended as a threshold to initiate treatment.23 Improvement in PbrO2 in response to increased CPP, blood transfusion,24 initiation of barbiturate coma,25 and after decompressive hemicraniectomy have all been observed.26 However, only retrospective observational studies have examined the effects of PbrO2 monitoring on outcomes after neurologic injury, with variable results reported.27,28 This may in part be due to the fact that PbrO2 monitors are very focused in their sampling territory, and data obtained from a monitor placed in a nonrepresentative area of the cortex may not be generalizable to the rest of the brain. A phase II, multicenter trial of PbrO2-guided therapy of patients with TBI (BOOST-2) is currently enrolling since 2011 and will hopefully provide more information about the influence of this monitoring modality on patient outcomes.
The device monitors a very local area of the brain tissue, and this technique is increasingly used for evaluation of cerebral oxygenation (normal PbrO2 values: 25 to 30 mm Hg).19 Monitoring may be performed in relatively undamaged parts of the brain or, preferably, in the penumbra region of an intracerebral lesion.20 Various studies have shown that an increase in ICP and a decrease in CPP or arterial oxygenation, and hyperventilation may result in decreased PbrO2. This monitoring modality has been shown to be helpful in detecting intraoperative brain hypoxia during episodes of hyperventilation.21 In patients with TBI, ischemic episodes defined as PbrO2 <10 mm Hg for longer than 15 minutes in the first week after the injury were found to be associated with unfavorable neurologic outcome, and values of 0 (not responding to an oxygen challenge) are compatible with brain death.22 A PbrO2 of 15 mm Hg or less has been recommended as a threshold to initiate treatment.23 Improvement in PbrO2 in response to increased CPP, blood transfusion,24 initiation of barbiturate coma,25 and after decompressive hemicraniectomy have all been observed.26 However, only retrospective observational studies have examined the effects of PbrO2 monitoring on outcomes after neurologic injury, with variable results reported.27,28 This may in part be due to the fact that PbrO2 monitors are very focused in their sampling territory, and data obtained from a monitor placed in a nonrepresentative area of the cortex may not be generalizable to the rest of the brain. A phase II, multicenter trial of PbrO2-guided therapy of patients with TBI (BOOST-2) is currently enrolling since 2011 and will hopefully provide more information about the influence of this monitoring modality on patient outcomes.
Microdialysis
Microdialysis uses a probe as an interface to the brain to continuously monitor the chemistry of a small focal volume of the cerebral extracellular space. This method uses internally perfused semipermeable membrane probes, which allow neurochemical water-soluble substances to be collected outside the brain for further analysis. Like PbrO2 monitors, the catheter is typically placed in a cerebral territory deemed to be at risk for further damage. In patients with SAH this is the vascular territory associated with the hemorrhage, in nonfocal TBI it is commonly the right frontal lobe, and in focal TBI and ischemic stroke it is commonly the penumbra region.29 The catheter allows measurement of energy-related metabolites, neurotransmitters, inflammatory markers, and exogenous substances such as drugs. The most commonly measured substances in clinical practice are lactate, pyruvate, glucose, and glutamate.29 This monitoring modality thus provides insight into the bioenergetic status of the brain. Increased lactate, decreased glucose, and an elevated lactate/pyruvate ratio indicate accelerated anaerobic metabolism. This metabolic pattern commonly occurs with cerebral ischemia or hypoxia, and increased glycolysis in this setting is associated with a poor outcome. Extracellular excitatory amino acids such as glutamate may provide a marker for secondary brain insults, as indicated by elevation during periods of hypoxia and intracranial hypertension. Like PbrO2 monitors, microdialysis catheters must be correctly positioned for the information they produce to be useful. Further, metabolism can be altered without changes of cerebral oxygenation and may not correlate with high ICP or low CPP, and correlation between microdialysis values and outcome is lacking.30
Diagnosis and Clinical Management of the Most Common Types of Neurologic Failure
Traumatic Brain Injury
TBI is the leading cause of death from blunt trauma, with an incidence of approximately 10/100,000 per year. With a proportion of 20% of deaths occurring in patients between the age of 5 and 45 years, TBI represents the leading cause of death in this age group. The most powerful predictors of poor outcome from injury through resuscitation are age >55 years, poor pupillary reactivity, postresuscitation Glasgow Coma Scale (GCS), hypotension, hypoxia, and an unfavorable intracranial diagnosis as established by radiologic features (e.g., CT scan). In addition, early hyperglycemia (>200 mg/dL) is a reliable independent predictor of poor outcome.
The GCS (see the chapter on Burns and Trauma) is the most widely used clinical measure of injury severity in patients with TBI. The advantages of this scale are that it provides an objective method of measuring consciousness, it has high intra- and interrater reliability across observers with a wide variety of experience, and it has an excellent correlation with outcome. However, the GCS is unmeasurable in up to 25% to 45% of the patients at admission and is inaccurate when only the partial score is used, such as in patients with tracheal intubation whose verbal response cannot be assessed. TBI qualifies as severe when the GCS is 8 or less. The predictive value of the GCS at admission is about 69% for good neurologic outcome and 76% for unfavorable outcome. After 7 days these figures approximate 80% for both favorable and unfavorable outcomes.31
Pupillary dilatation and light reactivity are also useful predictors of neurologic outcome. When both pupils are dilated and unreactive, the likelihood of poor neurologic outcome or death is as high as 90% to 95%. When both pupils are reactive, the likelihood of poor neurologic outcome is approximately 30% to 40%, while the probability of good outcome is 50% to 70%.
Hypotension is a strong predictor of poor outcome in TBI. Chesnut et al.32 reported that there was a 15-fold increased risk of mortality in patients with early hypotension and an 11-fold increase in mortality in patients with late hypotension.
Radiologic imaging is important in the diagnosis and in assessing the prognosis of patients with TBI. A number of computerized tomography (CT scan)–based scoring systems have been developed and correlated with outcome, but the amount of midline shift appears to be the single most predictive feature of outcome. Subarachnoid blood, intraventricular blood, and diffuse axonal injury patterns portend worse outcomes, while epidural hematomas generally have better outcomes.33 In addition, it should be noted that about one-third to one-half of the patients present no lesion at admission and develop new lesions secondarily, which is associated with substantially worse neurologic outcome.
The goal of resuscitation in TBI and other types of brain injury is to prevent continuing cerebral insult after a primary injury has already occurred. The extent of the primary cerebral injury is usually determined by the mechanism of the trauma, the cause, and the duration of cerebral ischemia. A primary insult is often associated with intracranial hypertension and systemic hypotension, leading to decreased cerebral perfusion and brain ischemia. Concomitant hypoxemia aggravates brain hypoxia, especially in the presence of hyperthermia, which increases brain metabolic demand. The combined effect of these factors leads to secondary brain injury characterized by excitotoxicity, oxidative stress, and inflammation. The resulting cerebral ischemia may be the single most important secondary event affecting outcome following a cerebral insult. Prevention of secondary injury is the main goal of resuscitative efforts.
TABLE 55-2. ICU MANAGEMENT OF PATIENTS WITH SEVERE TRAUMATIC BRAIN INJURY
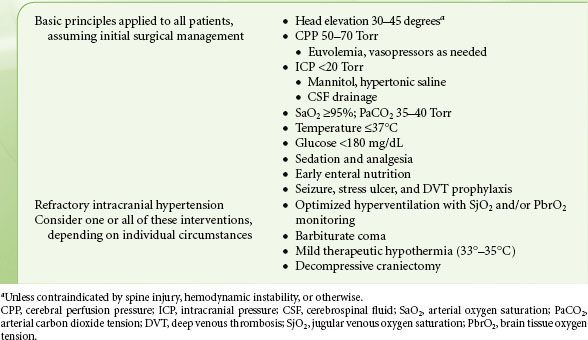
Traumatized areas of the brain manifest impaired autoregulation and disruption of the blood–brain barrier. If space-occupying lesions or edema are present, these will contribute to reduced brain compliance, leading to increased ICP and a consequent deleterious effect on cerebral blood flow. The rationale for attempting to optimize CPP arises from the fact that cerebral regions surrounding the primary lesion may be close to the ischemic threshold. Therefore, the goals of neuroresuscitation are oriented at restoration of cerebral blood flow by maintenance of adequate CPP, reduction of ICP, evacuation of space-occupying lesions, initiation of therapies for cerebral protection, and avoidance of hypoxia.
Unfortunately, the ICU treatment of TBI is hindered by a lack of rigorous RCTs to prove benefit, or lack thereof, for many of the management strategies utilized today. The Brain Trauma Foundation has published Guidelines for the Management of Severe Traumatic Brain Injury and revised them as recently as 2007.23 However, only one recommendation in 15 subject areas reaches “Level I,” meaning based on high-quality, randomized trials. Thus, treatment is based largely on pathophysiologic principles and uncontrolled trials. A general guideline for management of patients with severe TBI appears in Table 55-2. Basic principles of management of acute TBI, including osmotherapy, are discussed further in the chapter on Trauma and Burns; sedation, hyperventilation, hypothermia, corticosteroids, and antiseizure prophylaxis are discussed in further detail below.
Sedation of neurologically impaired patients should typically be achieved with short-acting sedatives to allow for frequent assessment of neurologic examination.34 Although no studies have investigated the effect of sedation on outcome in patients with neurologic disorders, a common practice is to provide sedation with propofol, benzodiazepines, or dexmedetomidine in patients following TBI. These agents have favorable effects on cerebral oxygen balance, although propofol is more potent in this regard. Undesirable effects of sedatives are those leading to a reduction in CPP due to hemodynamic depression or to an increase in CBF accompanied by a simultaneous increase in ICP, a condition potentially occurring, for example, with the use of ketamine.
Propofol rapidly penetrates the central nervous system and has rapid elimination kinetics. Despite the induction of systemic hypotension, propofol decreases cerebral metabolism resulting in a coupled decline in cerebral blood flow, with consequent decrease in ICP. Propofol’s favorable pharmacologic and neurophysiologic profile has led to its widespread use in neurointensive care, and high-dose propofol has been advocated as a substitute for barbiturate therapy in patients with refractory intracranial hypertension. However, prolonged (>24 hours), high-dose (>80 μg/kg/min) propofol administration has been associated with lactic acidosis, cardiac failure, and death (“propofol infusion syndrome”) in children and adults with TBI.35 Thus, the use of high-dose propofol to control refractory intracranial hypertension is not recommended, and barbiturates should be considered if ICP is not controlled by moderate doses of propofol.
The mechanisms by which barbiturates exert their cerebral protective effect appear to be mediated by a reduction in ICP via alteration in vascular tone, reduction of cerebral metabolic rate, and inhibition of free radical peroxidation. Although barbiturates are effective in reducing ICP, their routine use in TBI does not appear beneficial and may in fact result in excess mortality in patients with diffuse brain injury.36,37 This effect may in part relate to the profound cardiovascular depressant effects of barbiturates. Based on one small randomized trial, barbiturates do appear to reduce mortality in patients with refractory high ICP.38 Thus, high-dose barbiturate therapy may be considered in hemodynamically stable severe TBI patients with intracranial hypertension refractory to maximal medical and surgical ICP-lowering therapy. In some patients pentobarbital may induce cerebral hypoxia by reducing CBF in excess of metabolism, and therefore SjO2 monitoring may be considered during barbiturate therapy.
The centrally acting α-2 agonist dexmedetomidine has both sedative and analgesic effects. Its most desirable property is that it can allow for a more interactive and awake patient than other sedatives (see section on sedation later). While it has not been studied specifically in brain-injured patients, Drummond et al.39 have shown that cerebral blood flow–cerebral metabolic rate of oxygen (CMRO2) coupling remains intact in healthy volunteers during dexmedetomidine infusion.
Although neuromuscular blockade may result in a fall in ICP, the routine use of neuromuscular blockade is discouraged since its use has been associated with longer ICU course, a higher incidence of pneumonia, and a trend toward more frequent sepsis without any improvement in outcome.
Hyperventilation effectively reduces ICP by reducing CBF. However, the role that hyperventilation should play in routine management of TBI is not clear. Primarily, this is related to concerns that hyperventilation may lead to critically low CBF, resulting in worsening cerebral ischemia.40 In small randomized trials, prophylactic hyperventilation has not proven to be beneficial in TBI.41 In contrast, it has been proposed that “optimized hyperventilation” in the presence of “luxury perfusion” (excess CBF) may increase global cerebral oxygen metabolism and help normalize global cerebral glucose extraction. Cruz42 reported that an optimized hyperventilation strategy resulted in a reduction in mortality compared to CPP management in concurrent matched control patients, although this was not a randomized trial. On the basis of the available evidence, prolonged or prophylactic hyperventilation should be avoided after severe TBI, especially in the first 24 hours after the injury. Hyperventilation may be necessary for brief periods to reduce intracranial hypertension refractory to sedation, osmotic therapy, and CSF drainage and should be guided by SjO2 and/or PbrO2.23 A marked fall in either of these values suggests a harmful effect of hyperventilation and that it should be reduced or discontinued.
Experimentally, hypothermia causes a reduction in cerebral metabolism by decreasing all cell functions, both related to neuronal electric activity and those responsible for cellular integrity. In addition, mild hypothermia has been shown to decrease the release of substrates associated with tissue injury such as glutamate and aspartate. A meta-analysis of eight randomized trials of the use of mild hypothermia (33° to 35°C) in patients with TBI, and including 748 patients, indicated that despite a marginal improvement in poor neurologic outcome, there was no mortality advantage and there was an increased risk of pneumonia.43 An additional randomized trial of very early hypothermia published subsequently again found no outcome benefit to hypothermia.44 Therefore, mild hypothermia should not be routinely induced in patients with TBI, although it may be useful for rescue therapy of patients with refractory intracranial hypertension. On the other hand, immediate rewarming of TBI patients with spontaneous hypothermia may further worsen outcome and should be done with caution.45
 Corticosteroids to reduce posttraumatic inflammatory injury in TBI were advocated for 30 years or more, but without convincing evidence of benefit. The CRASH study, published in 2004, prospectively randomized over 10,000 patients presenting with acute TBI to receive high-dose methylprednisolone or placebo for 48 hours after hospital admission. Methylprednisolone administration was associated with an approximately 20% increase in the relative risk of death at 2 weeks in the entire cohort, and detriment was evident across subgroups divided by severity and type of injury.46 Thus, high-dose corticosteroids should not be administered as therapy for acute TBI.23 Likewise, intravenous magnesium administration did not improve outcomes in patients with TBI and might even have a negative effect in the treatment of significant brain injury.47
Corticosteroids to reduce posttraumatic inflammatory injury in TBI were advocated for 30 years or more, but without convincing evidence of benefit. The CRASH study, published in 2004, prospectively randomized over 10,000 patients presenting with acute TBI to receive high-dose methylprednisolone or placebo for 48 hours after hospital admission. Methylprednisolone administration was associated with an approximately 20% increase in the relative risk of death at 2 weeks in the entire cohort, and detriment was evident across subgroups divided by severity and type of injury.46 Thus, high-dose corticosteroids should not be administered as therapy for acute TBI.23 Likewise, intravenous magnesium administration did not improve outcomes in patients with TBI and might even have a negative effect in the treatment of significant brain injury.47
Anticonvulsants are effective at preventing early posttraumatic seizures within 7 days following head trauma. However, the evidence does not indicate that prevention of early seizures improves outcome following TBI.48 Seizures should therefore be treated on an as-needed basis.
Finally, it should be noted that use of albumin as fluid replacement therapy in patients with TBI has been associated with increased mortality in a subgroup analysis of an RCT comparing saline and albumin.49 Routine albumin administration to patients with TBI should be avoided (Table 55-2).
Subarachnoid Hemorrhage
The incidence of SAH in the United States varies from 7.5 to 12.1 cases per 100,000 population. SAH is most commonly caused by the rupture of an intracranial aneurysm. Other causes of SAH include trauma, vertebral and carotid artery dissection, dural and spinal arteriovenous malformations, mycotic aneurysms, sickle cell disease, cocaine abuse, coagulation disorders, and pituitary apoplexy. Aneurysmal SAH is associated with considerable morbidity and mortality, with only one-third of the patients suffering from SAH being functional survivors. The leading causes of death and disability are the direct effect of the initial bleed, cerebral vasospasm, and rebleeding. The Report of the Cooperative Study of Intracranial Aneurysms and SAH estimated that 33% of the patients would die before receiving medical attention.50,51 Although the number of early deaths may not have changed substantially, the overall case fatality rate of aneurysmal SAH has fallen over time, and although there is geographic variation mortality is reported in the 40% to 50% range in most studies.52 Approximately 40% to 50% of patients survive with good neurologic outcome after SAH (independence as measured by the modified Rankin scale).52 Severity of the initial bleed is the most important determinant of SAH outcome.
At the time of aneurysm rupture, there is a critical reduction in CBF due to increase in ICP toward arterial diastolic values. The persistence of a no-flow pattern is associated with acute vasospasm and swelling of perivascular astrocytes, neuronal cells, and capillary endothelium. After SAH, injury to the posterior hypothalamus may stimulate release of norepinephrine (NE) from the adrenal medulla and sympathetic cardiac efferent nerves. The release of NE has been associated with ischemic changes in the subendocardium (neurogenic stunned myocardium), cardiac dysrhythmias, and pulmonary edema.
In survivors of the initial bleed, emphasis has been placed on early aneurysm control with either surgery or interventional neuroradiology (coiling). Approximately 10% to 23% of unsecured aneurysms will rebleed in the first 2 weeks, most within the first 6 to 12 hours after the initial hemorrhage, and rebleeding is associated with mortality approximating 80%. Early aneurysm occlusion substantially reduces the risk of this complication. With the improvement of the operative management, delayed complications have become increasingly important causes of death and disability.
Cerebral vasospasm after SAH is identified by angiography in up to 60% of patients and is correlated with the amount and location of subarachnoid blood. A reduction in cerebral blood flow associated with vessel narrowing is ultimately responsible for the appearance of delayed ischemic neurologic deficits (DINDS). DINDS occur in approximately one-third of patients suffering from SAH. In a systematic review of the literature, Dorsch53 found an overall death rate of 31% (vs. 17% in patients without vasospasm), permanent deficits in 35%, and good outcome in 34% of the patients who developed symptomatic vasospasm. DINDS typically present as alteration in consciousness and/or transient focal neurologic deficits that rarely occur within the first 3 days after aneurysm rupture, typically peak in 7 to 10 days, and resolve over 10 to 14 days. If severe, vasospasm can result in cerebral infarction and persistent neurologic deficits, which contribute to considerable long-term morbidity.
TCD has been used to identify and quantify cerebral vasospasm on the basis that blood flow velocity increases as the diameter of the vessel decreases. Changes in measured velocities over time may be more reliable than absolute values in predicting symptomatic vasospasm. Velocities greater than 200 cm/s have been associated with a high risk of infarction but there is a poor correlation between the TCD velocities and angiographic findings, especially for the posterior circulation.
Oral nimodipine (60 mg every 4 hours for 21 days) as prophylaxis for cerebral vasospasm is recognized as an effective treatment in improving neurologic outcome (reduction of cerebral infarction and poor functional outcome) and mortality from cerebral vasospasm in patients suffering from SAH.54 Because angiographic studies did not demonstrate a difference in the frequency of vasospasm compared with a placebo-treated group, the benefits of nimodipine have been attributed to a cytoprotective effect related to the reduced availability of intracellular calcium and improved microvascular collateral flow. No other pharmacologic therapies to prevent or treat cerebral vasospasm have demonstrated effective results in clinical trials with the exception of statins. Tseng et al. conducted a meta-analysis of available studies examining the effect of statin therapy on outcomes in SAH and found a reduction in the risk of DINDS in those patients receiving a statin. Further, statin therapy appeared safe and may reduce the incidence of vasospasm although the quality of evidence was insufficient to conclude this definitively.55
Hypervolemic, hypertensive, and hemodilution (“triple-H”) therapy is one of the mainstays of prevention and treatment of cerebral ischemia associated with SAH-induced vasospasm despite the lack of evidence for its effectiveness, especially for prophylactic use.56 The rationale for hypervolemia is derived from the observation that hypovolemia is associated with poor outcomes after SAH and is often present because of blood loss and/or hypothalamic dysfunction and secretion of natriuretic peptides. Volume expansion is therefore considered beneficial to optimize the hemodynamic profile. The rationale for hypertension derives from the concept that a loss of cerebral autoregulation associated with vasospasm results in pressure-dependent cerebral blood flow. Finally, hemodilution is a consequence of hypervolemic therapy and is thought to optimize the rheologic properties of the blood and thereby to improve microcirculatory flow. There is no consensus with regard to the goals of the therapy and it is unclear which component of this therapy is necessary or sufficient to treat vasospasm.57 Common complications of treatment are pulmonary edema and myocardial ischemia. Since the blood–brain barrier may be disrupted, aggravation of vasogenic edema or hemorrhagic infarction has also been described.58
Interventional neuroradiology with the use of balloon angioplasty can reverse or improve vasospasm-induced neurologic deficits if initiated early after the development of ischemic symptoms. Although observational data suggest a beneficial effect of angioplasty on long-term outcomes, these findings have not been confirmed in RCTs, and the risks of angioplasty include intimal dissection, vessel rupture, ischemia, and infarction.
Hydrocephalus is another cause of neurologic dysfunction after SAH, occurring in 25% of patients surviving the hemorrhage. The presence of blood in the ventricular system obstructs ventricular drainage and CSF absorption sites (subarachnoid villi). Ventricular drainage is usually successful in improving neurologic symptoms due to hydrocephalus. A minority of patients will require a permanent ventriculo-peritoneal shunt. Seizures also occur in 13% of patients with SAH and are more common in patients with a neurologic deficit; thus, prophylactic anticonvulsant therapy should be considered, but consensus for its use is generally lacking.59
A relatively common complication after SAH (10% to 34%) is hyponatremia. Hyponatremia usually develops several days after the hemorrhage and is attributed to two main causes: (1) A syndrome of inappropriate antidiuretic hormone (SIADH), which is associated with euvolemia or mild hypervolemia and an excess of free water, and (2) cerebral “salt wasting,” which is marked by depletion of sodium and water. The differentiation of these two entities can be difficult but is theoretically important, in that SIADH is treated by free water restriction, and cerebral salt wasting with volume repletion and sodium administration. Thus, assessment of intravascular volume status is a key component when deciding on the treatment regimen for hyponatremia associated with SAH. Other medical complications are relatively common after SAH and include pneumonia, neurogenic pulmonary edema and acute lung injury (ALI), sepsis, gastrointestinal (GI) bleeding, deep venous thrombosis (DVT), and pulmonary embolism (PE).
Acute Ischemic Stroke
Although evidence indicates that the incidence of stroke has declined over the past 30 years, stroke remains one of the leading causes of disability and death in the United States. Nearly 90% of strokes can be attributed to an ischemic mechanism such as atherosclerosis, thrombosis, cardio-embolism, or hypotension. Other major causes of stroke are intracerebral hemorrhage and SAH.60 Unusual causes of stroke such as carotid artery dissection, hypercoagulation syndromes, or infective endocarditis should be considered in younger patients without apparent risk factors. Transient ischemic attacks may precede stroke and thus should be considered as a warning sign. The prognosis after stroke varies depending on the size and location of the lesion. In patients with acute ischemic stroke, the duration of coma appears to be the most important predictor of outcome and successful therapy.
Rapid clot lysis and restoration of circulation have been proposed as measures to limit the extent of brain injury and improve outcome after stroke. In accordance with the American Heart Association guidelines, systemic thrombolysis using IV alteplase (rt-PA) should be administered to patients presenting with acute ischemic stroke within 3 hours of symptom onset so long as no contraindications exist.61  A recent meta-analysis of trials including almost 3,700 patients found that the treatment window may be extended to 4.5 hours. In this analysis neurologic outcomes at 90 days in those patients treated with rt-PA within 4.5 hours of symptom onset were better than those treated with placebo, but no mortality benefit was found.62 After 4.5 hours the risk of hemorrhage (5% to 7% overall) appears to outweigh the benefits of treatment. It is clear that earlier treatment leads to improved outcomes and that treatment should be initiated as soon as possible.
A recent meta-analysis of trials including almost 3,700 patients found that the treatment window may be extended to 4.5 hours. In this analysis neurologic outcomes at 90 days in those patients treated with rt-PA within 4.5 hours of symptom onset were better than those treated with placebo, but no mortality benefit was found.62 After 4.5 hours the risk of hemorrhage (5% to 7% overall) appears to outweigh the benefits of treatment. It is clear that earlier treatment leads to improved outcomes and that treatment should be initiated as soon as possible.
There is no evidence that intra-arterial thrombolysis or mechanical thrombectomy is superior or inferior to intravenous administration of rt-PA, but they may be considered in patients with large MCA territory ischemic strokes and in situations where systemic rt-PA is contraindicated. Intravenous streptokinase, aspirin, heparin, warfarin, ticlopidine, or other antithrombotic or antiplatelet aggregating drugs are generally not recommended within 24 hours of treatment with either systemic or arterial rt-PA.
Unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH) have not been shown to prevent stroke progression or reduce the rate of stroke recurrence when administered within 48 hours of the acute event, and therefore their use is not recommended. In general, heparin is only recommended for early secondary prophylaxis in patients with suspected cardiac embolism. Aspirin 325 mg has been shown to reduce the risk of early recurrent ischemic stroke and is recommended within 24 to 48 hours of stroke onset in most patients, but increases the risk of hemorrhagic stroke. The frequency of DVT in acute stroke is reduced by anticoagulants, especially LMWH, but not by antiplatelet agents. However, it is unclear if the frequency of PE is also reduced.
The majority of patients with acute ischemic stroke present with severe arterial hypertension. If intracerebral hemorrhage is excluded, treatment of hypertension should be delayed since reduction of the perfusion pressure could compromise the viable brain surrounding the ischemia (ischemic penumbra). However, severe hypertension (systolic blood pressure >220 mm Hg, or mean arterial blood pressure of >130 mm Hg, or diastolic >120 mm Hg) should be controlled because of increased risk of hemorrhagic transformation in anticoagulated patients or after thrombolysis. Although there is no evidence for an optimal level of blood pressure, there is general consensus that the systolic pressure should not be lowered below 150 to 160 mm Hg. If the event is accompanied by raised ICP due to cerebral edema, the principles of treatment of raised ICP discussed above with regard to TBI similarly apply. Cytotoxic brain edema usually occurs 24 to 96 hours after acute ischemic stroke, and osmotherapy constitutes the basis of ICP reduction. Steroids are of no value in the treatment of ischemic stroke. Because hyperglycemia is associated with poor outcome in ischemic stroke, tight glucose control has been recommended by some. However, a randomized trial of glucose–potassium and insulin infusion that targeted normoglycemia in patients with acute stroke failed to demonstrate a mortality benefit at 90 days.63
Space-occupying middle cerebral artery infarctions (malignant MCA syndrome) and cerebellar infarctions have a high mortality rate and, in selected cases where signs of intractable intracranial hypertension are present, hemicraniectomy or decompressive surgery of the posterior fossa, respectively, could be life saving and improve outcome.64 Surgery seems to be less beneficial in patients presenting with aphasia, in patients older than 50 years, and in patients undergoing delayed surgery (greater than 24 hours after presentation).
Anoxic Brain Injury
Anoxic brain injury most commonly occurs as a result of cardiac arrest, either in or out of the hospital. Of patients who survive their initial cardiac arrest, in-hospital mortality ranges from approximately 50% to 90%, and a high percentage of survivors suffer brain injury with significant long-term disability. The pathophysiology of anoxic brain injury is multifactorial and includes excitatory neurotransmitter release, accumulation of intracellular calcium, and oxygen free radical generation. Unfortunately, pharmacologic therapies aimed at several of these pathways, including barbiturates, benzodiazepines, corticosteroids, calcium channel antagonists, and free radical scavengers, have failed to improve the outcome of anoxic brain injury.
 Mounting evidence supports a role for mild therapeutic hypothermia in anoxic brain injury. A Cochrane review in 2009 found that available evidence supports the induction of mild hypothermia (temperature 32° to 34°C) in survivors of out-of-hospital ventricular fibrillation/ventricular tachycardia arrest.65 Hypothermia was associated with a relative increase in patients with a favorable neurologic outcome (moderate disability or less) of 40% to 50%, and in one study it was also associated with relative 6-month mortality reduction by approximately 25%.66 Thus, mild therapeutic hypothermia should be routinely applied to comatose survivors of out-of-hospital cardiac arrest due to ventricular fibrillation; it should also be considered in other scenarios, including in-hospital cardiac arrest and cardiac arrest due to asystole and pulseless electrical activity.
Mounting evidence supports a role for mild therapeutic hypothermia in anoxic brain injury. A Cochrane review in 2009 found that available evidence supports the induction of mild hypothermia (temperature 32° to 34°C) in survivors of out-of-hospital ventricular fibrillation/ventricular tachycardia arrest.65 Hypothermia was associated with a relative increase in patients with a favorable neurologic outcome (moderate disability or less) of 40% to 50%, and in one study it was also associated with relative 6-month mortality reduction by approximately 25%.66 Thus, mild therapeutic hypothermia should be routinely applied to comatose survivors of out-of-hospital cardiac arrest due to ventricular fibrillation; it should also be considered in other scenarios, including in-hospital cardiac arrest and cardiac arrest due to asystole and pulseless electrical activity.
CARDIOVASCULAR AND HEMODYNAMIC ASPECTS OF CRITICAL CARE
Principles of Monitoring and Resuscitation
Shock states are associated with impairment of adequate oxygen delivery resulting in decreased tissue perfusion and tissue hypoxia. It is important to emphasize that global hemodynamic monitoring may not reflect regional perfusion or the peripheral tissue energy status. Occasionally, despite increased cardiac output (CO) and oxygen delivery peripheral tissues suffer from hypoxia due to blood flow maldistribution and uncoupling between oxygen delivery and oxygen utilization from mitochondrial dysfunction and energetic failure (cytopathic hypoxia).
Invasive monitoring in shock states provides insight into the circulatory status, organ perfusion, tissue microcirculation, and cellular metabolic status of the critically ill patient. Hemodynamic monitoring ranges from the simple monitoring of ECG and pulse oximetry, to continuous arterial pressure measurement via an arterial catheter, the monitoring of cardiac filling pressures with central venous or pulmonary artery catheters (PACs), to cardiac echocardiography. Several experimental monitoring devices detecting microenvironmental conditions at the tissue level are under continuous investigation.
Functional Hemodynamic Monitoring
 Pulmonary Artery Catheter
Pulmonary Artery Catheter
The PAC measures hemodynamic indices including central venous pressure (CVP), pulmonary artery pressure and occlusion pressure (PaOP), CO (thermodilution method), systemic vascular resistance (SVR) and pulmonary vascular resistance, and mixed venous oxygen saturation (SvO2) and provides data for deriving oxygenation variables (oxygen delivery [DO2], consumption [VO2], and extraction [O2ER]). The information provided by the PAC may assist in the differentiation of cardiogenic and noncardiogenic circulatory and respiratory failure and help guide fluid, inotropic, and vasopressor therapy. The technical and physiologic principles of the PAC are discussed in detail in other chapters.
The information provided by the PAC may assist in the differentiation of cardiogenic and noncardiogenic circulatory and respiratory failure and help guide fluid, inotropic, and vasopressor therapy. The technical and physiologic principles of the PAC are discussed in detail in other chapters.
Despite the theoretical benefits of pulmonary artery catheterization, there are few data to support a positive effect of PAC utilization on mortality or other substantive outcome variables. Patient populations including those with ARDS, congestive heart failure, and septic shock and high-risk surgical patients have all been the subject of investigations targeting the effect of PAC use on outcome.67,68 These trials have all failed to show benefit. As a consequence of these studies questioning the benefits of the PAC, the frequency of its use has substantially decreased in both medical and surgical patients.69
There is reason to believe that some or all of the following factors may be responsible for the observed lack of benefit associated with PAC use: (1) Device or procedure-related complications; (2) inaccurate data; (3) fundamentally incorrect assumptions about the meaning of the measured data; (4) inappropriate decisions resulting from misinterpretation of the data; and (5) harmful effects of well-intended therapies. As an example of factor number 3, there is increasing evidence that CVP and PA pressure do not predict the hemodynamic response to intravenous fluid administration in normal subjects or patients with shock.70,71 As an example of factor number 5, the ability to increase DO2 with fluid resuscitation and inotropic therapy in patients with septic shock identifies a better prognosis. This observation led to a therapeutic strategy known as “supraphysiologic resuscitation” to defined end points (cardiac index >4.5 L/min, DO2 >600 mL/m2/min, and VO2 >170 mL/m2/min) in patients with septic and surgical/trauma-related shock. However, a large, randomized, prospective study found that this approach was associated with increased mortality in patients with septic shock, and this approach is no longer favored.72
As with all interventions, the decision to insert a PAC must weigh the risks of monitoring against the potential benefits in terms of adaptation of treatment to the information obtained from the monitoring. While there are likely specific patient populations and disease states that may benefit from this monitoring modality, further research is necessary to define these groups.
A somewhat less invasive and less costly alternative to placing a PAC for the measurement of SvO2 is to measure central venous oxygen saturation (ScvO2) via a fiberoptic central venous catheter. ScvO2 is approximately 5 mm Hg higher than SvO2 in critically ill patients, but appears to correlate well with SvO2 during changes in hemodynamic status.73 Targeting an ScvO2 >70% as a component of early goal–directed therapy in patients with septic shock has been associated with a reduction in mortality.74
Arterial Pressure Waveform Analysis
In addition to static pressure measurements such as CVP and PaOP, dynamic indicators of preload include respiratory variation in systolic pressure and pulse pressure, both of which can be derived from the analysis of the waveform generated by a peripherally placed arterial catheter. In addition, techniques for deriving stroke volume, CO, and intrathoracic blood volume are available. These techniques present a less invasive approach to hemodynamic monitoring of critically ill patients.
In addition, techniques for deriving stroke volume, CO, and intrathoracic blood volume are available. These techniques present a less invasive approach to hemodynamic monitoring of critically ill patients.
The variation in systolic blood pressure and pulse pressure during positive pressure ventilation (PPV) is highly predictive of the response to intravascular fluid administration in both normal subjects and critically ill patients. During PPV there is an inspiratory reduction in right ventricular stroke volume due to decreased venous return and a subsequent reduction in left ventricular end-diastolic volume appearing during the expiratory phase of the respiratory cycle. Therefore, the left ventricular stroke volume varies cyclically with ventilation and is paralleled by a similar variation in systolic blood pressure and pulse pressure; these effects are exaggerated during absolute and relative hypovolemia. Systolic and pulse pressure variations are superior predictors of fluid responsiveness (compared to static measures such as CVP and PaOP) in patients with a variety of critical illnesses, including septic shock, ALI, and following cardiac surgery.75
Analysis of the systemic arterial pulse contour allows derivation of CO after initial calibration using an indicator-dilution technique. Two commercially available devices utilize either lithium (injected through a peripheral IV; LiDCO) or thermal dilution (injected through a central venous catheter; PiCCO) for initial calibration. CO derived using pulse contour analysis correlates well with thermodilution CO in a variety of conditions and has the advantage of providing continuous measurement without necessitating the placement of a PAC. The PiCCO device also allows for measurement of intrathoracic blood volume using transpulmonary thermodilution; the latter may be a more accurate reflection of preload than static central pressure measurements. Although further validation of these techniques in critically ill patients is necessary, the use of pulse contour analysis may potentially obviate the need for pulmonary artery catheterization to measure CO, particularly if combined with the measurement of ScvO2 as an indicator of the balance between oxygen delivery and consumption.
Transesophageal Doppler sonography using a small probe in a large nasogastric tube (6 mm) monitors descending aortic flow velocity continuously. When combined with the cross-sectional area of the aorta it allows for less invasive monitoring of CO compared to the PAC. The disadvantages of this technique are that it is inaccurate if not positioned correctly, it is easily dislodged, and that it does not measure the supra-aortic output or the aortic cross-sectional area directly. Instead, aortic cross-sectional area is determined using a nomogram and assumed to remain constant throughout systole.76 Two cases of intrabronchial displacement of the probe have been reported.
Echocardiography
Echocardiography is an even less invasive hemodynamic monitoring tool, see Chapter 26. Both transthoracic echocardiography (TTE), and transesophageal echocardiography (TEE) provide accurate diagnostic information with regard to dynamic ventricular function, valvular anatomy, pericardial anatomy and intracardiac pressures. While TEE has long been used in the ICU, especially in cardiac surgical patients, TTE use is becoming more widespread as the size and cost of the equipment are coming down. The major limitation of echocardiography is that it does not provide continuous monitoring and requires a high standard of training and experience. However, a number of studies have now shown that with very limited training practitioners can accurately and rapidly perform focused cardiac examinations. Melamed et al. provided intensivists with 2 hours of formal didactic training followed by 4 hours of hands-on training in focused point-of-care echocardiography. Using formal TTEs obtained by professional sonographers and interpreted by attending cardiologists as a gold standard, the intensivists were able to correctly identify ventricular (dys)function more than 80% of the time.77 Ultrasound has the additional benefit of being easily adapted to other uses in addition to cardiac assessment such as to guide placement of vascular catheters, assessment of lung pleura and parenchyma, and venous thrombosis detection. Although the role of echocardiography in routine critical care management remains undefined, it has been suggested that this technique can replace pulmonary arterial catheterization in the ICU without adversely affecting outcomes.78 However, it should be kept in mind that data on outcomes with the use of focused echocardiography are lacking.
Definition and Types of Circulatory Failure
The common denominator of shock is circulatory instability characterized by severe hypotension and inadequate tissue perfusion. Shock states are classified according to the primary cause of circulatory failure. Distributive or vasodilatory shock results from a reduction in SVR, often associated with an increased CO, whereas cardiogenic (left or right cardiac failure) and hypovolemic shock are low CO states usually characterized by increased peripheral resistance. Obstructive shock, as the name suggests, is characterized by an obstruction to CO such as may happen with a tension pneumothorax or pericardial tamponade. The most common forms of shock encountered in the ICU are cardiogenic, septic (a form of distributive shock), and hypovolemic shock. Despite extensive research and aggressive management, the mortality from shock remains staggeringly high; approximately 35% to 40% of patients die within 28 days of the onset of septic shock, and the mortality rate is 70% to 80% for patients with cardiogenic shock. The mortality from hypovolemic shock is highly variable and depends upon the etiology and the rapidity of recognition and treatment. Cardiogenic and septic shock are discussed in more detail in the following section; the causes and treatment of traumatic shock, including hypovolemic and obstructive shock, are discussed in the chapter on Trauma and Burns.
Cardiogenic Shock
The initiating event in cardiogenic shock is a primary pump failure. Heart failure may result from extensive myocardial infarction (MI), cardiomyopathy, dysrhythmias, mechanical complications (mitral regurgitation, ventricular septal defect), tamponade, and so on. The pathophysiologic characteristics include a reduction in contractility, usually accompanied by dilatation of cardiac cavities and venous congestion. Determining the etiology of cardiogenic shock is of utmost importance as the treatment varies considerably based on the underlying mechanism; β-blockers are contraindicated in nonischemic ventricular failure exacerbations and diuresis is frequently indicated, whereas in acute ischemic ST elevation MI (STEMI) β-blockers are a mainstay of treatment and there is little role for acute diuresis. A 12-lead ECG, serum troponin measurements, B-type natriuretic peptide levels, and TTE should be obtained in all patients presenting with heart failure as this information is helpful in differentiating between the various causes of heart failure.79 Absence of pulmonary congestion at initial clinical evaluation does not exclude a diagnosis of cardiogenic shock due to predominant left ventricular failure and is not associated with a better prognosis. The onset of pump failure is associated with two compensatory mechanisms: A reflex vasoconstriction in systemic vessels causing an increase in left ventricular workload and myocardial oxygen demand, and a redistribution of blood volume toward the heart and the lungs. However, cardiogenic shock developing within 36 hours of an acute MI has been associated with variably decreased SVRs, possibly mediated by the presence of a systemic inflammatory response.
Several studies have demonstrated that the incidence and severity of left ventricular failure complicating acute MI were directly related to the extent of ventricular mass necrosis. Consequently, therapy should minimize myocardial oxygen demand and raise oxygen delivery to the ischemic area; this goal is complicated by the fact that many resuscitative approaches to correct hypotension (preload augmentation, inotropes, and vasopressors; see later) increase myocardial oxygen consumption. In patients without hypotension, pharmacologic vasodilatation using nitrates or sodium nitroprusside may reduce myocardial oxygen consumption and improve ventricular ejection by reducing left ventricular afterload and possibly produce a shift of blood from the lungs to the periphery by reducing venous tone. B-type natriuretic peptide (nesiritide) and fenoldopam have similar effects, but have not shown any benefit in large randomized trials and are not recommended.80 When pharmacologic interventions are not sufficient to restore hemodynamic stability, the use of mechanical support with the insertion of intra-aortic balloon pump counterpulsation and ventricular assist devices can help unload the ventricles.
In patients with MI, coronary reperfusion can be achieved with systemic thrombolysis or, preferably, primary percutaneous coronary intervention (PCI). The American Heart Association/American College of Cardiology guidelines recommend treatment of STEMI by PCI within 90 minutes of presentation or, if not available, by thrombolytic therapy within 30 minutes of presentation.81 A randomized trial comparing emergency revascularization with primary PCI or coronary artery bypass surgery to a regimen of thrombolysis did not demonstrate a mortality difference at 30 days. However, at 6 months and 1 year, there was a significant mortality reduction with emergency revascularization in patients under age of 75 years.82 The addition of intra-aortic balloon counterpulsation to PCI in acute STEMI did not reduce infarct size or otherwise improve outcomes.83
Septic Shock
Septic shock is a form of distributive shock associated with the activation of the systemic inflammatory response and is usually characterized by a high CO, low SVR, hypotension, and regional blood flow redistribution, resulting in tissue hypoperfusion. Other causes of distributive shock include acute spinal cord injury, pancreatitis, burns, fulminant hepatic failure, multiple traumatic injuries, toxic shock syndrome, anaphylaxis and anaphylactoid reactions, and drug or toxin reactions, including insect bites, transfusion reactions, and heavy metal poisoning. In patients with systemic infections, the physiologic response can be staged on a continuum from a systemic inflammatory response syndrome (SIRS), to sepsis, severe sepsis, and septic shock (Table 55-3). The hemodynamic profile of septic shock is influenced by several sepsis-induced physiologic changes including hypovolemia and vasodilatation, in addition to cardiac depression. Sepsis is associated with a global decrease in cardiac contractility, and echocardiographic measurements of the left ventricle size demonstrate an inability of the ventricle to dilate in septic patients.84
In endotoxemia and sepsis, metabolic needs are increased and the ability of the tissues to extract and utilize oxygen may be impaired. Thus, a metabolic acidosis may be present despite normal levels of oxygen transport. A decrease in cellular O2 extraction capacity may result from factors other than hypoperfusion, such as direct cellular damage by toxins and/or mediators or maldistribution of blood flow (cytopathic hypoxia). The impact of impaired perfusion on organ function depends on individual susceptibility to hypoxia. The peculiar anatomy and the microcirculatory structure of intestinal villi with their countercurrent flow mechanism render the superficial layers of the mucosa particularly vulnerable to ischemia. The GI tract has been implicated as the “motor” of multiple organ dysfunction syndrome (MODS) and “splanchnic resuscitation” has been advocated as a central objective in patients with septic shock. Intramucosal pH and PCO2 monitoring has been proposed as a technique to assess the splanchnic metabolic state.
Though hypoperfusion is the dominant cause of lactic acidosis in sepsis, various degrees of intermediary metabolic alterations may contribute to the increased lactate production independent of perfusion. Normally, lactate is cleared by the liver metabolic activity via the Cori cycle. Subsequently, with the development of liver perfusion impairment this organ may turn to a net lactate producer. Furthermore, the increase in the rate of glucose metabolism may also occur due to inhibition of the step-limiting enzyme, pyruvate dehydrogenase, for pyruvate to enter the Krebs cycle. An increase in the relative proportion of inactive to active enzyme results in pyruvate accumulation and lactate production.
MODS refers to the presence of altered organ function in an acutely ill patient such that homeostasis cannot be maintained without intervention.85 The exact pathophysiology of MODS is not yet fully understood, although alterations in systemic hemodynamics, organ perfusion, and tissue microcirculation resulting in tissue hypoxia play a role in initiating and maintaining the syndrome. MODS accounts for most deaths in the ICU.
While organ failure only qualifies a dichotomous event that is either present or absent, organ dysfunction represents a continuum of physiologic derangements. Different severity scores have been proposed to quantify the range of severity of MODS. A commonly used score that correlates with a higher mortality in the ICU was developed by Marshall et al.86 This scoring system assigns increasingly high values based on markers of increasing respiratory, renal, cardiovascular, hepatic, hematologic, and central nervous system dysfunction.
TABLE 55-3. DEFINITIONS OF SEPSIS AND ORGAN FAILURE






Full access? Get Clinical Tree


