FIGURE 33.1 Mechanisms of bicarbonate reabsorption and secretion in the renal collecting tubules. CA = carbonic anhydrase, Pn = pendrin (the chloride-bicarbonate exchanger). See text for explanation.
Bicarbonate Secretion
Bicarbonate can also be secreted from specialized cells in the collecting ducts. This does not occur normally, but appears when there is excess HCO3 in plasma (i.e., metabolic alkalosis). The mechanism for HCO3 secretion is the reverse of HCO3 reabsorption, with the H+ pump on the capillary side of the tubular cells (see Figure 33.1). The principal difference is the presence of a chloride-bicarbonate exchange protein, called pendrin, on the luminal surface of the cells. This anion exchange protein has an important role in HCO3 secretion (5–8). The pendrin gene is up-regulated in metabolic alkalosis (6), and pendrin activity is directly related to the luminal chloride concentration (7).
Chloride Depletion
Chloride depletion plays a major role in promoting metabolic alkalosis by increasing HCO3 reabsorption and inhibiting HCO3 secretion. Both effects are mediated by a decrease in luminal chloride concentration in the distal nephron, which: (a) increases HCO3 reabsorption by promoting the movement of chloride and H+ into the tubular lumen, and (b) impairs HCO3 secretion by reducing the activity of the anion exchange protein, pendrin. The effects of luminal chloride on HCO3 secretion are considered the principal mechanism for the ability of chloride depletion to promote metabolic alkalosis, and for the ability of chloride replacement to correct metabolic alkalosis (5).
Hypokalemia
Like chloride depletion, hypokalemia promotes metabolic alkalosis by increasing HCO3 reabsorption and decreasing HCO3 secretion in the distal nephron. The increase in HCO3 reabsorption is caused by the simultaneous movement of K+ out of cells and H+ into cells, and the resulting decrease in intracellular pH promotes HCO3 reabsorption in the distal nephron (4). The decrease in HCO3 secretion in hypokalemia is attributed to a decrease in pendrin activity (8).
Aldosterone
Aldosterone (a mineralocorticoid produced in the adrenal cortex) promotes HCO3 reabsorption in the distal nephron by stimulating the membrane H+ pump on the luminal surface of acid-secreting renal tubular cells (9).
Predisposing Conditions
The three principal causes of sustained metabolic alkalosis are chloride depletion, hypokalemia, and mineralocorticoid excess. The clinical conditions that produce these derangements in ICU patients are described next.
Volume Loss
A decrease in extracellular volume promotes metabolic alkalosis. Early descriptions of this effect led to the term “contraction alkalosis,” but this term is misleading because the underlying mechanism is chloride depletion (i.e., the alkalosis is not corrected by replacing the volume deficit without chloride replacement, but it is corrected with chloride replacement without correcting the volume deficit) (5).
Loss of Gastric Acid
Nasogastric suction results in loss of gastric secretions, which are rich in H+ (50–100 mEq/L), CL– (120–160 mEq/L), and to a lesser degree, K+ (10–15 mEq/L)(10). Loss of H+ will initiate a metabolic alkalosis, while loss of CL– and K+ will sustain the alkalosis.
Diuretics
Thiazide diuretics and “loop” diuretics like furosemide promote metabolic alkalosis via chloride and potassium depletion. The principal action of these diuretics is to increase sodium loss in urine (natriuresis), and an equivalent amount of chloride is also lost in urine (chloruresis) because urinary chloride excretion usually matches urinary sodium excretion.
The increased luminal sodium also promote potassium loss in urine via the Na+– K+ exchange pump on the luminal surface of specialized cells in the distal renal tubules.
CLINICAL MANIFESTATIONS
Metabolic alkalosis has no apparent deleterious effects in most patients. In one case report, an elderly patient with protracted vomiting and a plasma [HCO3] of 151 mEq/L showed no signs of an immediate threat to life, and recovered fully after correction of the alkalosis (11). The lack of clinical consequences raises questions about the value of recognition and management of metabolic alkalosis.
Neurologic Manifestations
The neurologic manifestations attributed to alkalosis include depressed consciousness, generalized seizures, paresthesias, and carpopedal spasms. However, these manifestations are usually associated with respiratory alkalosis, not metabolic alkalosis. This is traditionally explained by the greater tendency for respiratory alkalosis to influence intracellular and central nervous system pH.
Neurologic manifestations seem to be more prominent in cases of metabolic alkalosis due to baking soda (sodium bicarbonate) ingestion (12), but this condition is accompanied by other electrolyte abnormalities (particularly hypercalcemia) that could be the responsible factor.
Hypoventilation
The ventilatory response to metabolic alkalosis is hypoventilation, with a subsequent rise in arterial PCO2. However, this is not a vigorous response, and a considerable rise in plasma HCO3 may be necessary before significant hypoventilation is evident (13). The ventilatory response to metabolic alkalosis is described by the following equation (14).
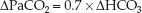 (33.1)
(33.1)
(PaCO2 is the arterial PCO2 and HCO3 is the plasma bicarbonate concentration.) This equation was used to construct the curve in Figure 33.2 that shows the relationship between PaCO2 and plasma HCO3 in progressive metabolic alkalosis. (A normal PaCO2 of 40 mm Hg and a normal plasma HCO3 of 24 mEq/L were used in constructing this curve.) Note that hypercapnia (i.e., PaCO2 >46 mm Hg ) does not occur until the plasma HCO3 increases from 24 to 32 mEq/L, which represents a 40% increase in plasma HCO3. In patients who have an increased ventilatory drive from an acute pulmonary process (e.g., pneumonia), even greater increases in plasma HCO3 will be necessary to produce significant hypoventilation.
Oxyhemoglobin Dissociation Curve
Alkalosis shifts the oxyhemoglobin dissociation curve to the left (Bohr effect), which results in a decreased tendency for hemoglobin to release oxygen into the tissues. When the O2 extraction from capillary blood is constant, a leftward shift of the oxyhemoglobin dissociation curve results in a decrease in venous PO2 (15), which typically indicates a decrease in tissue PO2. However, there is no evidence of inadequate tissue oxygenation (e.g., lactate accumulation) from this effect.
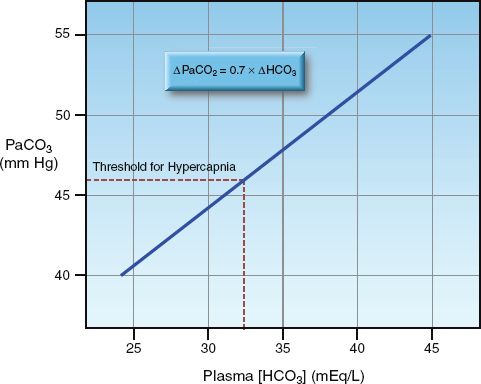
FIGURE 33.2 The relationship between plasma bicarbonate (HCO3) and arterial PCO2 (PaCO2) in metabolic alkalosis, as predicted by the equation shown at the top of the graph (using a normal PaCO2 of 40 mm Hg and a normal plasma [HCO3] of 24 mEq/L).
EVALUATION
The likely source(s) of metabolic alkalosis in ICU patients are loss of gastric secretions, diuretics, volume depletion, and hypokalemia, and these are readily apparent. In the rare case of uncertainty, the urinary chloride concentration can be informative, as described next.
Classification
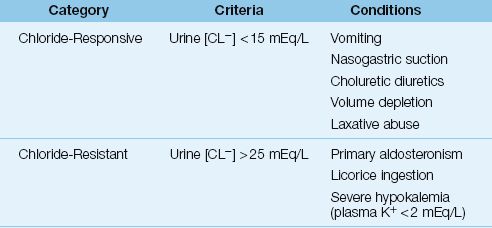
The urinary chloride concentration is used to classify metabolic alkalosis as chloride-responsive or chloride-resistant, and the conditions associated with each category are listed in Table 33.1.
Chloride-Responsive Alkalosis
A chloride-responsive metabolic alkalosis is characterized by a low urinary chloride concentration (i.e., <15 mEq/L), indicating chloride depletion. The principal conditions that produce this type of metabolic alkalosis include loss of gastric secretions, therapy with chloruretic diuretics (i.e., diuretics that promote urinary chloride excretion), and volume depletion. In cases of metabolic alkalosis due to chloruretic diuretics (e.g., thiazides or furosemide), the urinary chloride may be inappropriately high when the drug is active, but this effect is lost when the drug effect dissipates.
Table 33.1 Classification of Metabolic Alkalosis
Chloride-responsive metabolic alkalosis is typically accompanied by volume depletion, and improves with infusion of isotonic saline. Almost all cases of metabolic alkalosis in ICU patients are chloride-responsive.
LAXATIVE ABUSE: Diarrhea usually produces a hyperchloremic metabolic acidosis from bicarbonate losses in stool, but the diarrhea associated with chronic laxative abuse is rich in potassium and chloride (70–90 mEq/L), and it typically results in hypokalemia and a saline (chloride)-responsive metabolic alkalosis (16). The diagnosis can be elusive because patients often deny both laxative abuse and diarrhea.
Chloride-Resistant Alkalosis
A chloride-resistant metabolic alkalosis is characterized by an elevated urinary chloride concentration (i.e., >25 mEq/L). Most cases of chloride-resistant alkalosis are caused by primary mineralocorticoid excess (e.g., primary aldosteronism). Whereas volume depletion is common in chloride-responsive metabolic alkalosis, volume expansion is common in chloride-resistant metabolic alkalosis. As a result, this type of metabolic alkalosis does not improve with infusions of isotonic saline. This type of metabolic alkalosis is infrequent in ICU patients.
SEVERE K+ DEPLETION: Mineralocorticoid excess promotes urinary potassium loss, so hypokalemia is common in chloride-resistant metabolic alkalosis. However, a chloride-resistant metabolic alkalosis has been reported in patients with hypokalemia that is not caused by mineralocorticoid excess (17). The hypokalemia is typically severe (plasma K+ <2 mEq/L), and the proposed mechanism is diminished chloride reabsorption is the distal renal tubules. This condition is saline-resistant, but it can be corrected with potassium repletion (17).
MANAGEMENT
Since maintenance of a metabolic alkalosis is primarily the result of chloride and potassium depletion, these ions must be replenished to correct the alkalosis. This is accomplished with isotonic saline (0.9% NaCL) and potassium chloride.
Saline Infusion
Because volume depletion is common in chloride-responsive metabolic alkalosis, infusion of isotonic saline will help to correct the alkalosis. The volume of isotonic saline needed to correct the alkalosis can be estimated by first estimating the chloride (CL–) deficit, as shown below (2,18):
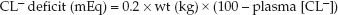 (33.2)
(33.2)
(wt is lean body weight in kg, and 100 is the desired plasma chloride concentration). The required volume of isotonic saline (in liters) is then estimated as follows:
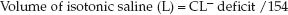 (33.3)
(33.3)
(154 is the chloride concentration in mEq/L in isotonic saline). This method is summarized in Table 33.2. If the patient is not hemodynamically compromised, rapid saline infusion is not necessary, and a rate of 100–125 mL/hr above total hourly fluid losses (including insensible loss) is appropriate.
Table 33.2 Saline Infusions for Metabolic Alkalosis

EXAMPLE: A 70 kg adult with protracted vomiting has a metabolic alkalosis with a plasma chloride of 80 mEq/L. The chloride deficit in this case (using a normal plasma [CL–] of 100 mEq/L) is 0.2×70×(100 – 80) = 280 mEq. The volume of isotonic saline needed to correct this deficit is 280/154 = 1.8 liters.
Edematous States
Isotonic saline infusion is contraindicated in edematous states because crystalloid fluids are distributed primarily in the interstitial fluid compartment. For every liter of isotonic saline infused, 825 mL will be added to the interstitial edema fluid (see Figure 12.2 on page 220) (19). Metabolic alkalosis in edematous states is often accompanied by hypokalemia, so potassium chloride replacement therapy can be used to treat the alkalosis.
Potassium Chloride
Potassium replacement is used to correct hypokalemia, and is always given as KCL, to take advantage of chloride replacement for correcting metabolic alkalosis. It is important to emphasize that diuretic-induced hypokalemia can be resistant to potassium replacement if there is concurrent magnesium depletion (8). Since magnesium depletion is also common during diuretic therapy, the plasma magnesium level should be checked at the outset of K+ replacement. (The evaluation of magnesium depletion is described in Chapter 37.)
Saline-Resistant Alkalosis
Alkalosis from mineralocorticoid excess (primary or secondary) is associated with an increased extracellular volume (which may be apparent as peripheral edema), and saline infusions will be counterproductive. Hypokalemia is common in this type of metabolic alkalosis, so K+ replacement (as KCL) helps in correcting the alkalosis. However, corrective therapy may require one of the following measures.
Acetazolamide
Acetazolamide (Diamox) inhibits carbonic anhydrase, the enzyme involved in HCO3 reabsorption and secretion (see Figure 33.1). The major effect of acetazolamide is inhibition of HCO3 reabsorption (proximal and distal sites) and an increase in urinary HCO3 excretion (the urine pH should rise to 7 or higher during acetazolamide therapy). The in-crease in HCO3 excretion is accompanied by an increase in sodium excretion, so acetazolamide provides the dual benefit of diuresis and correction of the metabolic alkalosis. The recommended dose is 5 to 10 mg/kg IV (or PO), and the maximum effect occurs after an average of 15 hours (21).
Table 33.3 Hydrochloric Acid Infusions

Hydrochloric Acid Infusion
Alkalosis that is not corrected with K+ replacement and acetazolamide can be treated with an infusion of dilute hydrochloric acid (HCL), but this can be risky, and is reserved only for severe alkalemia (plasma pH >7.5).
METHOD: The “dose” of HCL is determined by estimating the hydrogen ion (H+) deficit with the equation below (see also Table 33.3).
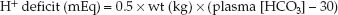 (33.4)
(33.4)
(wt is the lean body weight in kg, and 30 is the desired plasma [HCO3], which is higher than normal because the goal is to curb the alkalosis and not correct it). The preferred HCL solution for intravenous use is 0.1N HCL, which contains 100 mEq H+ and 100 mEq CL in one liter of distilled water. The volume of 0.1N HCL (in liters) needed to correct the H+ deficit is determined by the ratio (H+ deficit/100), as shown in Table 33.3. Because HCL solutions are corrosive, they should be infused through a large, central vein (22). and the infusion rate should not exceed 0.2 mEq/kg/hr (18).
EXAMPLE: Consider a lean, 70 kg adult with a severe, refractory metabolic alkalosis resulting in a plasma HCO3 of 40 mEq/L and an arterial pH of 7.57. The H+ deficit is 0.5×70×(40 – 30) = 350 mEq. The corresponding volume of 0.1N HCL is 350/100 = 3.5 L, and the maximum infusion rate is (0.2×70) = 0.14 L/hour (2.3 mL/minute).
ADVERSE EFFECTS: The major concern with HCL infusions is the corrosive effect on blood vessels. Extravasation of HCL solutions can produce severe tissue necrosis (and a fatal outcome), even when the solution is infused through a central vein (23).
A FINAL WORD
It’s Chloride
The final word in metabolic alkalosis is chloride, the principal element in both the maintenance and correction of metabolic alkalosis in ICU patients. Chloride plays an important role in metabolic acidosis as well. In addition to its role in acid-base balance, chloride is the second most abundant electrolyte in the extracellular fluid, and is a major factor in determining the osmolality and volume of extracellular fluid. In light of chloride’s participation in numerous physiological processes, a recent review of chloride suggested that it is the “queen of electrolytes” (24). Although a more appropriate term might be warranted, it highlights an increasing awareness that chloride is much more than sodium’s passive partner in the extracellular fluid.
REFERENCES
Reviews
1. Laski ME, Sabitini S. Metabolic alkalosis, bedside and bench. Semin Nephrol 2006; 26:404–421.
2. Khanna A, Kurtzman NA. Metabolic alkalosis. Respir Care 2001; 46:354–365.
3. Galla JH. Metabolic alkalosis. J Am Soc Nephrol 2000; 11:360–375.
Pathogenesis
4. Rose BD, Post TW. Regulation of acid-base balance. In: Clinical Physiology of Acid-Base and Electrolyte Disorders. 5th ed. New York: McGraw-Hill, 2001:325–371.
5. Luke RG, Galla JH. It is chloride depletion alkalosis, not contraction alkalosis. J Am Soc Nephrol 2012; 23:204–207.
6. Adler L, Efrati E, Zelikovic I. Molecular mechanisms of epithelial cell-specific expression and regulation of the human anion exchanger (pendrin) gene. Am J Physiol Cell Physiol 2008; 294L:C1261–C1276.
7. Vallet M, Picard N, Loffling-Cueni D, et al. Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol 2006; 17:2153–2163.
8. Wagner CA, Finberg KE, Stehberger PA, et al. Regulation of the expression of the CL/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int 2002; 62:2109–2117.
9. Winter C, Kampik NB, Vedovelli L, et al. Aldosterone stimulates vacuolar (H+-ATPase activity in acid-secreting intercalated cells mainly via a protein kinase C-dependent pathway. Am J Physiol Cell Physiol 2011; 1:C1251–C1261.
10. Gennari FJ, Weise WJ. Acid-base disturbances in gastrointestinal disease. Clin J Am Soc Nephrol 2008; 3:1861–1868.
Clinical Manifestations
11. Giovanni I, Greco F, Chiarla C, et al. Exceptional nonfatal metabolic alkalosis (blood base excess + 48 mEq/L). Intensive Care Med 2005; 31:166–167.
12. Fitzgibbons LJ, Snoey ER. Severe metabolic alkalosis due to baking soda ingestion: case reports of two patients with unsuspected antacid overdose. J Emerg Med 1999; 17:57–61.
13. Javaheri S, Kazemi H. Metabolic alkalosis and hypoventilation in humans. Am Rev Respir Dis 1987; 136:1011–1016.
14. Adrogue HJ, Madias NE. Secondary responses to altered acid-base status: The rules of engagement. J Am Soc Nephrol 2010; 21:920–923.
15. Nunn JF. Nunn’s Applied Respiratory Physiology. 4th ed. Oxford: Butterworth-Heinemann Ltd, 1993:275–276.
Evaluation
16. Roerig JL, Steffen KJ, Mitchell JE, Zunker C. Laxative abuse. Epidemiology, diagnosis, and management. Drugs 2010; 70:1487–1503.
17. Garella S, Chazan JA, Cohen JJ. Saline-resistant metabolic alkalosis or “chloride-wasting nephropathy”. Report of four patients with severe potassium depletion. Ann Intern Med 1970; 73:31–38.
Management
18. Androgue HJ, Madias N. Management of life-threatening acid-base disorders. Part 2. N Engl J Med 1998; 338:107–111.
19. Imm A, Carlson RW. Fluid resuscitation in circulatory shock. Crit Care Clin 1993; 9:313-333.
20. Whang R, Flink EB, Dyckner T, et al. Mg depletion as a cause of refractory potassium depletion. Arch Intern Med 1985; 145:1686–1689.
21. Marik PE, Kussman BD, Lipman J, Kraus P. Acetazolamide in the treatment of metabolic alkalosis in critically ill patients. Heart Lung 1991; 20:455–458.
22. Brimioulle S, Vincent JL, Dufaye P, et al. Hydrochloric acid infusion for treatment of metabolic alkalosis: effects on acid-base balance and oxygenation. Crit Care Med 1985; 13:738–742.
23. Buchanan IB, Campbell BT, Peck MD, Cairns BA. Chest wall necrosis and death secondary to hydrochloric acid infusion for metabolic alkalosis. South Med J 2005; 98:822.
A Final Word
24. Berend K, van Hulsteijn LH, Gans RO. Chloride: the queen of electrolytes? Eur J Intern Med 2012; 23:203–211.







Full access? Get Clinical Tree


