Edited by Mark Little Ian Rogers Heat-related disorders have a broad range of potential aetiologies and manifestations. In some, the primary disorder is a failure of thermal homoeostasis whereas, in others, the hyperthermia is secondary to other processes. The major heat-related illnesses to consider are exercise-associated collapse (EAC), heatstroke, neuroleptic malignant syndrome, serotonin toxicity and malignant hyperthermia. Although of different aetiologies, the latter four, all associated with significantly elevated core temperatures, share much common ground with regard to complications and treatment. EAC is the most common heat-related illness presenting either to medical tents at sporting events or to emergency departments. EAC manifests at the end of a race when muscle pump enhanced venous return ceases and cardiac output drops. This leads to collapse, often with a brief loss of consciousness. Despite the claims of the advocates of fluid loading and sports drinks in exercise, the primary mechanism is a failure of prompt baroreceptor responses and not haemodynamically significant dehydration. Severe heat-related dehydration is rare. The other, more serious, heat-related disorders are all associated with, or the potential for, significant hyperthermia which, if not treated promptly, results in similar pathophysiology at a cellular and organ system level. A core body temperature around or greater than 41.5°C results in progressive denaturing of a number of vital cellular proteins, failure of vital energy-producing processes and loss of cell membrane function. At a cellular level, the exact mechanisms leading to loss of cell membrane function and cell death in heat illness remain uncertain. At an organ system level, these changes may manifest as rhabdomyolysis, acute pulmonary oedema, disseminated intravascular coagulation, cardiovascular dysfunction, electrolyte disturbance, renal failure, liver failure and permanent neurological damage [1,2]. Any or all of these complications must be expected in severe heat illness. The hallmark of heatstroke is failure of the hypothalamic thermostat, leading to hyperthermia and the associated additional pathophysiological features described above. Clinically, heatstroke can be divided into ‘exertional heatstroke’ due to exercise in a thermally stressful environment, and ‘classic heatstroke’, which occurs in patients with impaired thermostatic mechanisms. Common risk factors for heatstroke are listed in Table 28.1.1. Table 28.1.1 Behavioural Army recruits Athletes Exertion Inappropriate clothing Elderly Inappropriate exposure Babies left in cars Manual workers Pilgrims Drugs Anticholinergics Diuretics Phenothiazines Salicylates Stimulants/hallucinogens Illness Delirium tremens Dystonias Infections Seizures Certain drugs produce hyperthermia by mechanisms in addition to interference with thermostatic function. In severe serotonin toxicity and neuroleptic malignant syndrome, increased motor activity and central resetting of the hypothalamic thermostat combine to produce hyperthermia. In the case of serotonin toxicity, these effects are a consequence of a relative excess of central nervous system serotonin whereas, in neuroleptic malignant syndrome, dopamine depletion or dopamine receptor blockade is responsible. The elevation of central nervous system serotonin in serotonin toxicity is usually associated with combinations of serotoninergically active drugs, taken either therapeutically or in overdose. The incidence of serotonin toxicity when such combinations are taken is not known, but is low and there is much individual variation in susceptibility [3]. Less often the syndrome is precipitated by a single serotoninergic agent. Drugs associated with the serotonin syndrome are listed in Table 28.1.2. The most commonly implicated are combinations of serotinergically active drugs, such as selective serotonin reuptake inhibitors (SSRIs), lithium, pethidine, monoamine oxidase inhibotors (MAOIs) and amphetamines. Table 28.1.2 Drugs causing severe serotonin toxicity Antidepressants Monoamine oxidase inhibitors (MAOIs) Selective serotonin reuptake inhibitors (SSRIs) Selective serotonin and noradrenaline reuptake inhibitors (SSNRIs) St John’s wort Tricyclics Analgesics Pethidine Tramadol Recreational drugs Amphetamines Methylenedioxymethamphetamine (MDMA, ‘ecstasy’) Neuroleptic malignant syndrome (NMS) is a rare idiosyncratic reaction to neuroleptic agents with an incidence of between 0.02% and 3.0%, depending on the diagnostic criteria used. It occurs in response to a single agent, usually at therapeutic dosage. In individuals, the occurrence may be dose related. Certain at-risk groups have been identified and are listed in Table 28.1.3. Table 28.1.3 Risk factors for neuroleptic malignant syndrome Patient factors Agitation Dehydration Male sex (male:female=2:1) Organic brain disease Drug dosing factors Depot neuroleptics High initial neuroleptic dose High-potency neuroleptic (e.g. haloperidol) Rapid dosage increase NB: Duration of drug exposure and toxic overdose are not related to risk of developing NMS. Malignant hyperthermia is a genetically inherited disorder in which triggering agents cause a release of sarcoplasmic Ca2+ stores. The resulting elevation of myoplasmic Ca2+ stimulates many intercellular processes, including glycolysis, muscle contraction and an uncoupling of oxidative phosphorylation. This leads to hyperthermia that, in contrast to neuroleptic malignant and serotonin syndromes, is purely peripheral in origin. Prevention of exertional heatstroke should focus on the education of at-risk groups. Dehydration is not as important aetiologically in heatstroke as once thought. Exertional heatstroke is most often reported in shorter, high intensity exercise where marked dehydration is unlikely. So, although adequate fluid intake is needed for prolonged exercise, it is not a key factor in heatstroke prevention. As high ambient temperatures and high humidity predispose to exertional heatstroke, exertion in these environments should be limited. The clinical presentation of EAC will be familiar to all emergency practitioners as it mirrors that of poor cerebral perfusion from any other cause. Patients complain of nausea, vomiting, malaise and dizziness. There may be a history of collapse and there is likely to be a tachycardia and (orthostatic) hypotension. The orthostatic hypotension may manifest at the end of physical exertion by collapse with brief loss of consciousness that is typical of EAC. In this syndrome, and in distinction to heatstroke, the core temperature will be less than 40°C and neurological function will rapidly return to normal once the patient is lying down. The classic clinical features of heatstroke are neurological dysfunction, core temperature above 41.5°C and hot, dry skin. However, relying on this classic triad to make the diagnosis will result in a number of cases being missed. Loss of consciousness is a constant feature of heatstroke [1] but, by the time of ED presentation, conscious state may be improving, although some neurological abnormality will persist. Temperature readings may be misleadingly low, due either to effective pre-hospital care or to measurements at inappropriate sites, such as the oral cavity or axilla. Profuse sweating is a much more common feature than previously thought [1]. Other clinical features may include tachycardia, hyperventilation, seizures, vomiting and hypotension. Serotonin syndrome is characterized by CNS, autonomic and motor dysfunction (Table 28.1.4). It is a clinical diagnosis. It develops after a latent period, which is normally a few hours, but may be as long as several days. The spectrum of illness produced is broad. Most patients are only mildly affected and may escape clinical detection. Only the most serious develop hyperthermia (usually in the setting of muscular rigidity) severe enough to produce the complications of rhabdomyolysis, disseminated intravascular coagulation and renal failure. Most cases will resolve within 24–48 h once the precipitating agents are withdrawn. Even in severe cases, the underlying biochemical abnormality rapidly improves, usually with the institution of muscular paralysis. The morbidity and mortality in these cases is caused by the complications that develop while the syndrome is active. Table 28.1.4 Features of the serotonin toxicity Central nervous system Agitation Anxiety Confusion Decreased level of consciousness Seizures Motor Clonus Hyperreflexia Hypertonia Incoordination Myoclonus Tremor Autonomic Diaphoresis Diarrhoea Hypertension Hyperthermia Tachycardia This syndrome manifests in patients who have recently been started on neuroleptic treatment or in whom the dose of a neuroleptic agent has been increased. It has been associated with almost all antipsychotics (both first and second generation) and has also been reported in patients in whom a dopaminergic agent has been rapidly withdrawn (e.g. in parkinsonism). There is a latent period of many hours to several days. Characteristically, there are four classic signs: fever, rigidity, altered mental state and autonomic instability. In practice, it may be difficult to distinguish clinically from serotonin toxicity unless a good drug history is obtained. As in serotonin toxicity, the spectrum of illness is very broad, with only the more severe cases developing hyperthermia and its complications [5]. This occurs when a triggering agent is given to a susceptible individual, usually in the context of an anaesthetic. Triggering agents identified include inhalational anaesthetic agents, such as halothane, isoflurane and enflurane, as well as succinylcholine and ketamine. The first signs are failure to achieve muscle relaxation following succinylcholine, tachypnoea and tachycardia. If not recognized and treated, acidosis, rhabdomyolysis and hyperthermia will ensue. In some cases, signs and symptoms may be delayed or even reappear after apparently successful treatment, so that malignant hyperthermia may even present as a postoperative fever. Untreated, the mortality is as high as 70%, but this can be reduced tenfold by appropriate management. Diagnosis of the hyperthermic disorders is based on the history, clinical picture and exclusion of alternative diagnoses. Investigations are thus directed towards excluding other possible causes of temperature elevation (e.g. infection, metabolic disorders) and evaluation of the specific complications of hyperthermia. Patients with a presumed clinical diagnosis of EAC should still have serum electrolytes and creatine kinase measured to exclude exercise associated hyponatraemia and rhabdomyolysis, respectively. Should mental state not rapidly normalize with supine posture, then an urgent finger prick or serum glucose estimation is needed. Collapsed athletes should also have an ECG to identify unrecognized cardiac abnormalities. All other heat disorders warrant a far more extensive laboratory and radiological work-up, as multiorgan system dysfunction is the rule [2,6]. Tests must include an ECG, serum electrolytes, disseminated intravascular coagulation (DIC) screen, liver function tests, muscle enzyme assays, renal function and urinalysis, serum glucose and a chest X-ray. EAC responds rapidly to supine posture (ideally with the legs elevated), rest and oral fluids. Intravenous normal saline is rarely required as few athletes will be profoundly dehydrated. The use of ‘routine’ IV normal saline in collapsed athletes should be actively discouraged as it will worsen exercise associated hyponatraemia where there are persistent, inappropriate antidiuretic hormone levels. This is a true medical emergency. Early recognition and aggressive therapy in the field and in hospital can prevent substantial morbidity and mortality. The key management is aggressive cooling. Cooling rates of at least 0.1°C/min should be achievable. Several cooling methods have been proposed, including evaporative cooling, iced water immersion, ice slush, cool water immersion, iced peritoneal lavage and pharmacological methods [7]. A combination of methods is most widely used in EDs. All of the patient’s clothing should be removed and the patient sprayed with a fine mist of tepid water while gentle fanning is commenced (a ceiling fan is ideal). At the same time, areas with vascular beds close to the surface (neck, axillae and groins) should be packed with ice bags. This technique facilitates patient access and monitoring when compared to methods, such as ice-bath immersion even though an iced bath may offer more rapid cooling. Although ice-cold IV fluids can also aid in rapid cooling (as used in therapeutic hypothermia post-cardiac arrest), fluid requirements in heatstroke can be difficult to estimate and balance. In hospital, shivering, seizures and muscle activity may need to be controlled with pharmacological agents, such as chlorpromazine, benzodiazepines and paralysing agents. Aspirin and paracetamol are ineffective and should be avoided. Intravenous fluids need to be used cautiously and may need titrating to central venous or pulmonary capillary wedge pressures. Maintain adequate oxygenation but avoid hyperoxia. Ventilatory support may be required. Urine flow needs to be maintained with initial volume loading, and later with mannitol or furosemide, to prevent secondary renal injury, especially from rhabdomyolysis. Electrolyte, acid–base and clotting disturbances should be closely monitored and treated by standard measures. Treatment of the drug-related hyperthermia involves both specific pharmacological therapy and full supportive and cooling measures, as described above. The objective is to recognize and treat before serious complications occur. In mild cases of the serotonin syndrome, no treatment or small doses of benzodiazepines may be all that is required while awaiting spontaneous resolution. In severe cases, neuromuscular paralysis should be considered early, especially in cases of markedly altered mental state. The duration of treatment used is partly judged on the half-life of the presumed causative agents [4]. Specific antiserotoninergic drugs that can be used include chlorpromazine (12.5–50 mg IM/IV) and cyproheptadine (4–8 mg orally 8-hourly). Again, early recognition and full supportive care, combined with specific therapy, is the mainstay of treatment. Dopamine agonists, such as bromocriptine, may reduce the duration of the syndrome. It can be administered orally or by nasogastric tube at an initial dose of 2.5–10 mg tds. Dantrolene acts by inhibiting the release of calcium from the sarcoplasmic reticulum and is the specific agent used in the treatment of malignant hyperthermia. It should be given in addition to full supportive care and discontinuing the triggering agents. The dose is 2.5 mg/kg IV initially, repeated every 15 min up to a maximum of 30 mg/kg if needed. In heatstroke, both the maximum core temperature and the duration of temperature elevation are predictors of outcome. Prolonged coma and oliguric renal failure are poor prognostic signs [1]. Mortality is still of the order of 10%, but most survivors will not suffer long-term sequelae [1,2]. Any patient with suspected heatstroke should routinely be referred to the intensive care unit for ongoing care. Most cases of EAC will be suitable for short-stay ED treatment or, indeed, simply for treatment on-site in an event medical tent. Prognosis in the drug-related group of hyperthermia is dependent largely on the degree to which the complications have progressed before definitive and aggressive treatment is begun. Again, early referral to intensive care is indicated. Even with appropriate treatment, mortality for malignant hyperthermia approaches 7%. After recovery, the patient’s medication regimen will need to be reassessed although, in the case of neuroleptic malignant syndrome, it may be possible slowly to reintroduce a neuroleptic agent at a lower dose. With malignant hyperthermia, future anaesthesia will need to be modified to avoid precipitating agents. In addition, family members should be tested for susceptibility. Ian Rogers Hypothermia is defined as a core temperature of less than 35°C. This can be measured at a number of sites (including oesophageal, right heart, tympanic and bladder). Rectal remains the routine in most emergency departments (EDs), despite concerns at how rapidly it equilibrates to and reflects true core temperature. Conventionally, hypothermia is divided into three groups: mild (32–35°C), moderate (29–32°C) and severe (<29°C) on the basis of measured core temperature. In a field setting, where core temperature measurements may not be possible, moderate and severe are often grouped together as they typically share the clinical features of absence of shivering and altered mental state. These categorization systems can be used both out of and in hospital as a guide to selecting rewarming therapies and prognosis. Mild hypothermia is considered the stage where thermogenesis is still possible; moderate is characterized by a progressive failure of thermogenesis; and severe by adoption of the temperature of the surrounding environment (poikilothermia) and an increasing risk of malignant cardiac arrhythmia. Nevertheless, there are substantial differences between individuals in their response to hypothermia. Hypothermia may occur in any setting or season [1]. True environmental hypothermia occurring in a healthy patient in an adverse physical environment is less common in clinical practice than that secondary to an underlying disorder. Common precipitants include injury, systemic illness, drug overdose and immersion and are outlined in more detail in Table 28.2.1. The elderly are at greater risk of hypothermia because of reduced metabolic heat production and impaired responses to a cold environment. Alcohol is a common aetiological factor and probably acts by a number of mechanisms, including cutaneous vasodilatation, altered behavioural responses, impaired shivering and hypothalamic dysfunction. Hypothermia in the ED setting is often associated with underlying infection [2]. Table 28.2.1 Despite substantial individual variations, it is still possible to describe the typical patient in each category of hypothermia. The clinical manifestations of hypothermia also depend on the features of the underlying aetiology. Mild hypothermia manifests clinically as shivering, apathy, ataxia, dysarthria and tachycardia. Moderate hypothermia is typically marked by a loss of shivering, altered mental state, muscular rigidity, bradycardia and hypotension. In severe hypothermia, signs of life may become almost undetectable, with coma, fixed and dilated pupils, areflexia and profound bradycardia and hypotension. The typical cardiac rhythm of severe hypothermia is slow atrial fibrillation. This may degenerate spontaneously, or with rough handling, into ventricular fibrillation or asystole. In the field, moderate and severe hypothermia are often grouped together, with the key clinical feature of absent shivering suggesting the loss of the ability to rewarm without medical intervention. Many complications may also manifest as part of a hypothermia presentation, although at times it may be difficult to separate cause from effect. These include cardiac arrhythmias, thromboembolism, rhabdomyolysis, renal failure, disseminated intravascular coagulation and pancreatitis. Mild hypothermia with shivering and without apparent underlying illness needs no investigation in the ED. Moderate or severe hypothermia mandates a comprehensive work-up to seek common precipitants and complications that may not be clinically apparent. Biochemical and haematological abnormalities are frequently associated with hypothermia [1], although there is no consistent pattern. Blood tests that are indicated include sodium, potassium, glucose, renal function, calcium, phosphate, magnesium, amylase, creatine kinase, ethanol, full blood count and clotting profile. Arterial blood gases, if taken, should be accepted at face value, rather than adjusting for the patient’s temperature. Impaired ciliary function, stasis of respiratory secretions or aspiration may be expected in moderate-to-severe hypothermia, so chest radiography should be routine. Other radiology may be indicated if a trauma-related aetiology is suspected. A 12-lead electrocardiograph and continuous ECG monitoring should be routine in moderate-to-severe hypothermia. The typical appearance is slow atrial fibrillation, with J or Osborn waves most prominent in leads II and V3–V6 (Fig. 28.2.1). The J wave is the extra positive deflection after the normal S wave and is more obvious and more commonly seen with increasing severity of hypothermia. The general and supportive management of hypothermia victims largely follows that of other critically ill patients. However, some syndrome-specific issues demand careful attention. Muscle glycogen is the substrate preferentially used by the body to generate heat by shivering. All hypothermics, therefore, need glucose. In mild cases, this can be given orally as sweetened drinks or easily palatable food. With more severe hypothermia, gastric stasis and ileus are common and glucose should be given intravenously: 5% dextrose can be infused at 200 mL/h. Additional volume resuscitation with normal saline or colloid should be gentle, bearing in mind the contracted intravascular space in severe hypothermia and that hypotension that would be classified as severe at a core temperature of 37°C is a normal physiological state at 27°C. All intravenous fluids should be warmed to minimize ongoing cooling. Endotracheal intubation by a skilled operator is safe in severe hypothermia. Intubation is indicated as in any other clinical condition to provide airway protection or to assist in ventilation. Ventilatory support and, where necessary, manipulation of acid–base status, should be titrated to maintain uncorrected blood gas pH and PCO2 within the normal range. The slow atrial fibrillation so common in more severe hypothermia is a benign rhythm and requires no chemical or electrical correction. It will revert spontaneously with rewarming. Pulseless ventricular tachycardia and ventricular fibrillation should largely be managed along conventional lines. However, if initial DC shocks are unsuccessful, then others are unlikely to be so until the patient is warmer. Repeat countershocks are generally reapplied with every 1°C increase in core temperature. Magnesium may be the antiarrhythmic drug of choice in hypothermia. The pharmacokinetics and dynamics of most drugs are substantially altered at low body core temperatures. Indeed, for many of the common drugs used in an ED they are unknown. Insulin is known to be inactive at<30°C. Hyperglycaemia, due in part to loss of insulin activity, is common in hypothermia, but should probably be managed expectantly until sufficient rewarming has occurred to ensure full endogenous insulin activity. Strategies for rewarming in hypothermia have only a limited evidence base on which to base recommendations. Although more invasive and rapid techniques are advocated for more severe hypothermia, there is little evidence to support this advice. The traditional concern of afterdrop (a paradoxical initial drop in core temperature with rewarming) is probably of little or no relevance in a clinical setting [3]. Rewarming therapies are broadly divided into three groups: endogenous rewarming, which is allowing the body to rewarm by its own endogenous heat production; external exogenous rewarming, which is supplying heat to the outside of the body; and core exogenous rewarming, which is applying the heat centrally. The classification of the commonly utilized rewarming therapies is outlined in Table 28.2.2. Table 28.2.2 Rewarming therapy classification Endogenous rewarming is a mandatory component of any emergency rewarming protocol. It consists of drying the patient, covering them with blankets, placing them in a warm and wind-free environment and warming any intravenous or oral fluids that are administered. Endogenous rewarming alone can be expected to rewarm at a rate of about 0.75°C/h. For most patients above 32°C (the level at which shivering thermogenesis is typically preserved), endogenous rewarming is the only therapy required. The exception is the exhausted patient in whom shivering has ceased at a core temperature higher than expected. Although more sophisticated techniques, such as bath immersion, will more rapidly rewarm a mildly hypothermic patient, there is no evidence that an increased rewarming rate improves prognosis in this group. In moderate hypothermia, endogenous heat production is likely to fail progressively and more aggressive exogenous rewarming therapies are indicated. Hot-bath immersion has the theoretical disadvantage of causing peripheral vasodilatation, with shunting of cool blood to the core and convective heat loss. This might be expected to increase core afterdrop and produce circulatory collapse. In fact, rewarming rates of at least 2.5°C/h with minimal afterdrop have been achieved using baths at 43°C [4]. Nevertheless, substantial practical difficulties are obvious with monitoring a more seriously ill patient immersed in a bath. This method of rewarming can only be recommended for otherwise healthy patients who are expected to make a rapid recovery from accidental environmental hypothermia (e.g. immersion in very cold water). The two therapies that have been best studied and are widely used in moderate hypothermia are forced-air rewarming and warm humidified inhalation [5]. Forced-air rewarming is achieved by covering the patient with a blanket filled with air at 43°C. These devices direct a continuous current of air over the patient’s skin through a series of slits in the patient surface of the blanket. This method produces minimal, if any, afterdrop, is apparently without complication and, combined with warm humidified inhalation, should produce rewarming at about 2.5°C/h. The value of warm humidified inhalation is probably by preventing ongoing respiratory heat loss. Given its widespread availability and lack of complications, it seems reasonable to combine it with forced-air rewarming in moderate hypothermia [6]. Body-to-body contact and chemical heat packs are often recommended as field treatments for all degrees of hypothermia. In mild hypothermia, it seems that the benefit of any heat they deliver is negated by an inhibition of shivering thermogenesis. In more severe cases, where shivering is absent, it may be that even the small amount of exogenous heat they deliver is beneficial, but this remains unproven. In severe hypothermia, more aggressive exogenous rewarming therapies may be indicated in order rapidly to achieve core temperature above 30°C, the threshold below which malignant cardiac arrhythmias may occur spontaneously. Bladder, gastric or peritoneal lavage with warm fluids are all relatively ineffective methods of heat transfer and, as such, are not recommended for use in emergency situations. When available, full cardiopulmonary bypass achieves rewarming rates of about 7.5°C/h without core afterdrop. Pleural lavage using large volumes of fluid warmed to 40–45°C through an intercostal catheter may be nearly as effective. Both techniques have the advantage of delivering heat to the heart which acts as a heat pump to distribute rewarming to key core organs, but are clearly invasive and carry associated risks. These risks are certainly acceptable in a hypothermic arrest but, in the non-arrested patient, a slower rate of rewarming using forced-air and warm humidified inhalation may be more appropriate. A suggested rewarming algorithm based on the evidence available to date is shown in Figure 28.2.2. Attempts at developing a valid outcome prediction model for hypothermia are likely to be frustrated by its multifactorial aetiology. Recovery with appropriate treatment is likely from accidental environmental hypothermia when there is no associated trauma. To date, the coldest patient to survive accidental hypothermia neurologically intact had an initial measured temperature of 13.7°C [7]. Although increasing severity of hypothermia does worsen prognosis, the major determinant of outcome is the precipitating illness or injury. Reported mortality rates vary from 0 to 85%. Mild hypothermics without associated illness or injury can be safely managed at home in the care of a responsible adult. Moderate hypothermia may be treatable in a short-stay observation ward, but often requires a longer inpatient stay to manage underlying illness or injury. Severe hypothermics are at risk of multiorgan system complications and should be considered for admission to an intensive care unit. Controversies David R Smart This chapter focuses on medical problems that develop secondary to breathing gases at higher than normal atmospheric pressure (dysbarism). This usually occurs in the context of scuba (self-contained underwater breathing apparatus) diving, a popular recreational activity in Australasia. Diving is generally very safe and serious decompression incidents occur approximately 1:10 000 dives. However, because of a high participation rate, between 200 and 300 cases of decompression illness are treated in Australia each year. It is estimated that 10 times that number of divers experience less serious health problems after diving. Emergency physicians are often the first medical staff to assess the diver after a diving accident and it is essential they understand the risks and potential injuries. An understanding of pressure and some gas laws is essential to understand the pathophysiology of diving injuries. The air pressure at sea level is 1 atmosphere absolute (ATA). Multiple units are used to measure pressure (Table 28.3.1). For every 10 m a diver descends in sea-water, the pressure increases by 1 ATA. This pressure change impacts on gas spaces within the body according to Boyle’s law. Table 28.3.1 Atmospheric pressure at sea level in various units 1 Atmosphere absolute (ATA) 101.3 kPa (SI units) 1.013 Bar 10 m of sea water (MSW) 760 mm of mercury (mmHg) 14.7 pounds per square inch (PSI) Boyle’s law states that, at a constant temperature, the volume of a gas varies inversely to the pressure acting on it: where P=pressure, V=volume and k=constant. The proportionate change in volume is greatest near the surface (Table 28.3.2). Table 28.3.2 Depth vs pressure and gas volume (Boyle’s law) Dalton’s law states that the total pressure (Pt) exerted by a mixture of gases is equal to the sum of the pressures of the constituent gases (Px, Py, Pz): Therefore, as divers breathe air at increasing atmospheric pressure, the partial pressures of nitrogen and oxygen increase: A diver breathing air at 40 m is inhaling a gas with a partial pressure of oxygen equivalent to breathing 100% oxygen at the surface. At partial pressures above 3 ATA, the PN2 affects coordination and judgement (‘nitrogen narcosis’). Oxygen may also become toxic at partial pressures greater than 1 ATA. Recreational scuba diving generally has a limit of 40 m because of these effects. Henry’s law states that at a constant temperature the amount of a gas that will dissolve in a liquid is proportional to the partial pressure of the gas in contact with the liquid: where Q=volume of gas dissolved in a liquid, k=constant and Pgas=partial pressure of the gas. Henry’s law is relevant in diving illness because it is the basis of decompression illness (DCI). As the ambient pressure increases, the diver is exposed to increasing partial pressures of nitrogen, which dissolves in bodily fluids. The amount of nitrogen absorbed depends on both the depth (which determines the partial pressure of nitrogen) and the duration of the dive. Tissues also take up nitrogen at different rates depending on their blood supply and permeability. Eventually, the tissues become saturated with nitrogen and no further absorption occurs. As the diver ascends and ambient pressure decreases, the partial pressure of nitrogen in some tissues will exceed ambient pressure, resulting in tissue supersaturation. If the diver ascends slowly enough, nitrogen diffuses out of the tissues and is transported, safely dissolved in the blood, to the lungs for elimination. This is known as ‘off-gassing’. If the diver ascends too rapidly, sufficient nitrogen bubbles will form in their body to cause decompression illness. Oxygen does not cause problems because it is rapidly metabolized by the tissues. Barotrauma occurs when changes in ambient pressure lead to expansion or contraction of gas within enclosed body cavities. The change in gas volume distorts or tears adjacent tissue. Injury by this mechanism may occur to the middle ear, inner ear, sinuses, lungs, eyes (via the diver’s mask) and, rarely, the gut. Different injury patterns occur in breath-hold divers (snorkellers) compared to those breathing compressed air. Both breath-hold and scuba divers may experience injury of the middle and inner ear, sinuses and eyes if they do not equalize pressures in the gas spaces as they descend. Breath-hold divers are unlikely to injure their lungs as their lung volumes reduce as they descend and return to their original volume as they ascend to the surface by the increasing ambient pressure. Middle-ear barotrauma (MEBT), the most common medical disorder of diving, usually occurs during descent. Increased ambient pressure results in a reduction of middle-ear volume. If equalization of the volume via the eustachian tube is inadequate, a series of pathological changes results. The tympanic membrane (TM) is deformed inwards, causing inflammation and haemorrhage. Middle-ear mucosal oedema is followed by vascular engorgement, effusion, haemorrhage and, rarely, TM rupture. Symptoms of middle-ear barotraumas include ear pain, tinnitus and conductive hearing loss. Mild vertigo may also be experienced. More severe vertigo and pain occur if water passes through a perforated TM. Severe vertigo and significant sensorineural hearing loss should alert the emergency physician to possible inner-ear barotrauma (IEBT) (see below). MEBT severity is graded by visual inspection of the TM (Table 28.3.3). An audiogram is useful to document any hearing loss. Table 28.3.3 Grading of severity of middle-ear barotrauma Treatment of MEBT consists of analgesia, decongestants and ear, nose and throat (ENT) referral if there is TM perforation or suspected IEBT. Antibiotics are indicated for TM rupture because of potential contamination with water. The patient should not dive again until symptoms and signs have resolved, any TM perforation has healed, and the eustachian tube is patent. Sudden pressure changes between the middle and inner ears can cause rupture of the round or oval windows or a tear of Reissner’s membrane. This usually occurs during rapid descent without equalizing or forceful Valsalva manoeuvres. Symptoms include sudden onset of tinnitus, vertigo, nausea and vomiting, vestibular symptoms and profound sensorineural hearing loss, which may not be apparent until the diver has left the water. Onset of symptoms after the dive while performing an activity that increases intracranial pressure (e.g. heavy lifting) suggests IEBT. Coexistent middle-ear barotrauma is absent in about one-third of cases. The main differential diagnosis is DCI involving the inner ear or vestibular apparatus. Inner-ear DCI usually occurs on deep dives using helium and oxygen mixtures (heliox) and is typically accompanied by other symptoms or signs of DCI. Frequently it is difficult to distinguish between IEBT and vestibular DCI. Isolated inner-ear DCI has been reported in sports divers breathing air. Treatment of IEBT consists of avoidance of activities that increase intracranial pressure and urgent (same day) ENT referral for more detailed assessment and audiometry. Surgical repair may be undertaken when vertiginous symptoms are severe. Vomiting should be treated with antiemetics and the diver kept supine with their head on a pillow. If DCI is excluded, then a 45° semirecumbent position is preferred. If DCI cannot be excluded the diver should have a trial of recompression. In one series, exposure to pressure did not worsen the diver’s condition. The benefit of steroids in IEBT has not been confirmed. It was thought that further diving was contraindicated after IEBT, but recent case data suggest that diving might be possible following full recovery of hearing. Ear-canal barotrauma is very rare and only occurs if there is a complete obstruction of the canal (usually by wax or ear plugs), creating a non-communicating gas cavity between the obstruction and the TM. Treatment is symptomatic. ENT specialist referral may be necessary if the TM cannot be visualized. Mucosal swelling and haemorrhage occur if the communication of the sinuses with the nasopharynx is blocked and equalization of sinus pressure is not possible during descent. The frontal sinuses are most commonly involved. Sinus pain usually develops during descent. Maxillary sinus involvement can refer pain to the upper teeth or cheek. There may be resolution of the pain at depth, due to mucosal oedema and blood filling the volume deficit left by gas compression. Pain and epistaxis may occur as the diver ascends. The pain usually persists after diving. Tenderness will be noted over the affected sinus. In doubtful cases, a sinus CT (computed tomography) scan will assist the diagnosis. Treatment includes analgesia, decongestants and recommendations to avoid diving until asymptomatic. Antibiotics may be required if secondary infection occurs. If divers fail to exhale air into their masks on descent, the reduced volume inside the mask can cause pain, petechiae and conjunctival haemorrhage. In assessing these divers, it is important to confirm that they have normal visual acuity and fundi are normal. Treatment is with analgesia alone. Expansion of gas within the gastrointestinal tract on ascent can occasionally cause colicky abdominal pain. Rupture of the stomach is rare but has occurred where panic or equipment failure has led to air swallowing and rapid ascent. Presentation is with abdominal pain and distension. Shoulder pain may be due to diaphragmatic irritation or coexisting DCI. Sub-diaphragmatic free air may be visible on an erect chest X-ray. The differential diagnosis includes pulmonary barotrauma, because air can enter the peritoneum via the mediastinum and oesophageal or aortic openings in the diaphragm. The diagnosis is confirmed with endoscopy and surgical repair is necessary. Severe tooth pain may occur with descent or ascent if air is trapped under a decaying tooth or recent filling. Percussion of the involved tooth is painful. Treatment is with analgesia and dental repair. Breathing compressed air at depth, the diver’s lungs contain greater amounts of gas than they would on the surface. Divers are trained to breathe continuously during ascent or to exhale continuously if they have lost their air supply. Pulmonary barotrauma results when a diver ascends without exhaling adequately and the expanding gas in the lungs exceeds the lung’s elasticity, tearing alveoli. This occurs most commonly when a diver runs out of air, panics and ascends too rapidly. The change in pressure over 1 m near the surface is sufficient to cause lung barotrauma. It has been reported in student divers training in swimming pools and in helicopter escape training. It can also occur with a normal ascent if there is a localized area of lung that does not empty properly, as is possible in divers with asthma, reduced pulmonary compliance or air trapping. The resultant clinical syndromes depend on the sites at which the air escapes and include arterial gas embolism (AGE), pneumomediastinum and pneumothorax. Onset of symptoms is usually rapid. If pneumomediastinum or pneumothorax is detected after diving, it is essential to look for features consistent with associated gas embolism. These include impairment or loss of consciousness, cognition impairment including loss of memory, or neurological abnormalities. Sometimes the abnormalities are subtle and tests of cognition and memory should be performed in addition to a detailed history and thorough examination. If AGE is suspected, then the affected individual should be kept supine and urgent recompression treatment is required. Management of AGE is discussed under the heading of decompression illness. Lung barotrauma is regarded as the cause of AGE, however, in early studies, only about 5% of divers with AGE had radiographic evidence of a pneumothorax on plain chest X-ray. Subtle signs of extra-alveolar air suggesting pulmonary barotrauma are present in nearly half with more sophisticated imaging, such as CT. The reverse also applies. If divers present with a pneumomediastinum or pneumothorax, then they may have up to 50% chance of AGE. The signs of AGE in these circumstances may be subtle with only a brief period of loss of memory or dizziness. Pneumomediastinum and subcutaneous emphysema can usually be managed conservatively. If symptoms are severe, 100% oxygen can accelerate resolution of the trapped gas. If recompression is required for coexistent AGE, then the pneumomediastinum does not require any specific additional management unless a pneumothorax is present. Isolated pneumothorax resulting from pulmonary barotrauma is very uncommon. Pneumothorax from pulmonary barotrauma should be managed in the same way as non-diving-related causes and recompression is not necessary. If recompression is required for coexisting AGE, a chest tube with a Heimlich valve should be placed before commencing treatment, because the size of any remaining pneumothorax will increase markedly on depressurization. Once the acute management of pneumomediastinum and pneumothorax has occurred, the divers should be referred to a diving medical specialist for long-term follow up, because the conditions will impact upon their future diving fitness. Diving accidents involving bubbles were traditionally divided into decompression sickness (DCS; due to nitrogen bubbles coming out of tissue) and arterial gas embolism (AGE; due to pulmonary barotrauma releasing air into the circulation). DCS is then classified as type I or II. Type I DCS involves the joints or skin only; type II involves all other pain, neurological injury, vestibular and pulmonary symptoms. In the 1990s, the term ‘decompression illness’ (DCI) was proposed to include both DCS and AGE, for the following reasons: The current classification system describes DCI in terms of four components: For example, a diver may be classified as having acute progressive neurological DCI with no evidence of barotrauma. The classification has been generally adopted in Australia and New Zealand, but not in North America. DCI is a satisfactory term from a management perspective but, from a scientific perspective, it does not describe differing aetiologies and pathophysiology. DCI occurs if excessive nitrogen comes out of solution to form bubbles which gain access to the venous and lymphatic systems or if bubbles form within tissues themselves. The formation of bubbles requires tissues to be supersaturated with nitrogen and for ascent to be excessively rapid. As bubbles form in tissues, they distort tissue architecture, which results in impaired function, pain and inflammation and is probably responsible for most musculoskeletal symptoms. Many bubbles entering the venous system do not cause symptoms. In fact, using ultrasonic detection methods, intravascular microbubbles are detected after approximately 60% of routine dives. It appears that these bubbles are safely filtered by the lung and diffuse into the alveoli. Bubbles entering the arterial system are more likely to cause serious problems. This can occur under several circumstances. Large volumes of bubbles may overwhelm the pulmonary filter and arterialize. Bubbles may also bypass the lungs via a right-to-left shunt. Up to one-quarter of the population may have a patent foramen ovale (PFO). Under normal circumstances, the valve over the foramen is kept closed by the pressure difference between the left and right atria. However, during diving, the pressure differential may reverse during a Valsalva manoeuvre or with acute increases in right-sided pressures associated with a large pulmonary gas load. Alternatively, gas can enter the circulation following pulmonary barotrauma. Air entering the pulmonary arterial system is carried to the pulmonary capillaries where it is trapped and reabsorbed by the alveoli. Air entering the pulmonary venous system, however, will pass through the heart and result in AGE. Gas bubbles entering the circulation (either from tissues or barotrauma) cause both mechanical and biochemical abnormalities. Trapping in the pulmonary circulation may result in elevation of right heart and pulmonary pressures, leading to increased venous pressures, reduced cardiac output and impairment of tissue microcirculation. Arterial bubbles can cause end- organ ischaemia, although most pass through the capillaries and into the venous system. Most of the deleterious effects are a consequence of secondary inflammation of the vascular endothelium. Bubble–endothelial interaction activates complement, kinin and coagulation systems and precipitates leucocyte adherence. This results in increased vascular permeability, interstitial oedema and microvascular sludging. The end result is ischaemia and haemoconcentration. Increased vascular permeability of the cerebral circulation will produce cerebral oedema. Vasospasm and reduced flow occurs approximately 1–2 h after bubbles have passed through the arterial tree. This explains the commonly observed clinical course of a diver with a cerebral AGE experiencing an initial deterioration (bubble emboli), followed by spontaneous improvement (bubbles pass through the cerebral capillaries) and then a subsequent secondary deterioration. Animal studies have demonstrated that bubbles travel against arterial flow because of their buoyancy and lodge in the highest point of the body (hence the logic of maintaining supine position after decompression accidents). A number of dive tables and computer algorithms have been developed in an attempt to avoid nitrogen supersaturation of tissues and improve diver safety. Limits are placed on depth, time and ascent rates to allow safe decompression after diving. However, as with all mathematical models which attempt to predict biological behaviour, the dive tables are far from perfect. One series has shown that 39% of DCI cases were within the limits of the table they were using and 24% within the limits of the conservative Canadian Defence and Civil Institute of Environmental Medicine (DCIEM) tables. Historically, it was assumed that DCI could not occur after dives shallower than 10 m, but it is now known that this can occur, particularly if there has been more than one dive per day or multiple ascents. The occurrence of DCI is a probabilistic event where risk increases with increasing depth, time, numbers of dives, numbers of ascents and rates of ascent. Flying after diving can precipitate DCI. Even if there are no bubbles at the end of the dive, excess nitrogen remains in the tissues and is slowly off-gassed. Further reduction in ambient pressure at altitude can cause bubbles or enlarge pre-existing asymptomatic ones. Current guidelines advise against flying for 12 h after a single short no-decompression dive and 24 h following multiple or decompression dives. Onset of any symptoms during or in the hours after diving should be regarded as DCI until proven otherwise. Failure to recognize and treat milder cases can lead to permanent morbidity because the disease can progress as the bubble load increases with time. Early onset of symptoms or signs (up to 1 h), especially those that are neurological in nature, indicates a serious decompression emergency and recompression is a time-critical treatment. Milder syndromes of decompression illness may develop up to 24 h after a dive or even later if there is a precipitant, such as heavy exercise or ascent to altitude (e.g. flying). In general, pulmonary barotrauma that results in AGE has a dramatic clinical presentation and the onset of major neurological symptoms and signs occurs within seconds to minutes after the dive. DCI caused by intravascular bubbles from barotrauma can be rapidly fatal and has a mortality of 5% in sport divers who reach a recompression chamber alive. In Australia, it is the second most common cause of diving-related death after drowning. The brain is the organ most commonly affected, probably because of the vertical positioning of the diver on ascent. Cerebral gas emboli can cause sudden loss of consciousness, convulsions, visual disturbances, deafness, cranial nerve palsies, memory disturbance and asymmetric multiplegias. Hemiplegia is much less common than asymmetric multiplegias. Symptoms almost always begin within 10 min of surfacing. Sudden loss of consciousness on surfacing should be assumed to be due to cerebral gas emboli. Spontaneous improvement may occur with first aid measures, but relapse is common. Coronary arterial emboli rarely may present as acute myocardial infarction or arrhythmia. Abdominal organs and skin may also be embolized. Elevation of serum creatine kinase (predominantly from skeletal muscle), serum transaminase and lactate dehydrogenase levels in divers with AGE suggests that emboli are distributed more extensively than previously recognized. Peak creatine kinase (CK) may be a marker of the degree and severity of AGE. Onset of DCI due to gas bubbles coming out of solution can be equally as dramatic (especially after rapid ascents from deep dives), but frequently evolves over hours post-dive. DCI caused by bubbles released from tissues usually causes symptoms within 1 h of completing a dive and 90% of cases have symptoms within 6 h. Neurological symptoms occurring around 30 min after a dive have been associated with PFO. Common symptoms include profound fatigue, myalgia, periarticular pain and headache. Shoulders and elbows are the joints most commonly involved. The pain is usually a dull ache, which may initially be intermittent and migrate from joint to joint, but later becomes constant. Movement aggravates the pain, but local pressure with an inflated sphygmomanometer cuff may improve it. Paraesthesia and numbness may accompany the pain suggesting concomitant neurological disease. Neurological DCI may present as personality change, headache, memory loss, visual defects, convulsions, confusion and altered level of consciousness. A flat affect may be the only symptom. The vestibular system can also be involved, with dizziness, vertigo, vomiting, nystagmus and ataxia. Spinal-cord involvement occurs in up to 60% of cases of neurological DCI. The exact cause of spinal DCI is still debated. It may be a result of venous infarction of the cord due to obstruction of the epidural vertebral venous plexus. Other explanations include ischaemia and inflammation from bubble emboli or the formation of local bubbles within the spinal cord (autochthonous bubbles). Symptoms include back pain, paraesthesia and paraplegia, with bowel and bladder involvement. It is potentially disastrous to misdiagnose back pain coming on a few minutes after a dive as musculoskeletal pain and not consider spinal cord DCI. If the bubble load overwhelms the pulmonary filter, a diver can present with a syndrome known as pulmonary DCI or ‘the chokes’. The symptoms of this syndrome include dyspnoea, pleuritic substernal chest pain, cough, pink frothy sputum, cyanosis and haemoptysis. It indicates the diver has sustained a large intravascular gas load, so a careful inquiry about other symptoms of DCI is mandatory and, if present, recompression is advised. Diving-related pulmonary oedema and salt-water aspiration syndrome are the major differential diagnoses. A variety of rashes may be caused by cutaneous bubbles; however, these syndromes affect less than 10% of divers. The most common presentations are pruritus with no rash, a scarlatinaform rash with pruritus and cutis marmorata. Cutis marmorata begins as a spreading erythema but subsequently develops a marbled appearance of pale areas surrounded by cyanotic mottling. The injured diver requires simultaneous assessment and treatment. One hundred per cent oxygen treatment should be continued during the assessment. If the history suggests AGE, the patient should be kept in the horizontal position to avoid re-embolization. If symptoms are progressing rapidly, the examination should be brief but thorough so as to ensure rapid access to recompression. In serious cases, some of the historical information may be obtained once the diver is receiving treatment in the recompression chamber. The diagnosis of DCI is made on history and examination. A full dive history must be obtained, in addition to the medical history. Important details include the number of dives over recent days, depth, bottom time (the time from beginning descent to beginning direct ascent), performance of any decompression or safety stops, dive complications, such as rapid ascents, surface interval between dives and the time interval between completing the dive and onset of symptoms. Previous dive experience, equipment used and gases breathed should also be recorded. A history of using surface supply equipment (the ‘Hookah’ apparatus) should alert the examining physician to the possibility of carbon monoxide poisoning and carboxyhaemoglobin measurement is required. Cold water, hard exercise during the dive, increasing age, multiple ascents and repetitive dives are predisposing factors in the development of DCI. Any exposure to altitude (>300 m) or heavy exercise post-dive should be recorded. A thorough examination, particularly of the neurological system, to detect subtle abnormalities is required. It is also helpful to perform basic tests of cognitive function, such as the mini-mental state examination. For milder static DCI syndromes with delayed presentation, the sharpened Romberg test provides useful information. It is performed by asking the patient to stand heel-to-toe with open palms on opposite shoulders. The patient is stable. They are then asked to close their eyes and timed until they lose balance or achieve 60 s. A score of less than 60 s is suggestive of DCI in an injured diver. This test should not be performed if the history was suggestive of AGE or if there are neurological symptoms or signs. Recompression should only be delayed for investigations if they will directly alter management. A full blood count and electrolytes are useful in that intravascular fluid depletion is common in severe DCI and the degree of haemoconcentration may correlate with eventual neurological outcome. Serum CK and liver function tests (LFTs) may be indicators of gas embolism; however, these do not influence clinical management. The blood glucose level should be checked in divers with impaired consciousness. A chest X-ray is indicated if pulmonary barotrauma is suspected, because a pneumothorax requires treatment before recompression. A dilemma occurs if the diver has a neurological presentation, because they should not be moved from the horizontal position until they are recompressed. If CAGE is suspected and CT is available, a supine CT scan of the thorax is preferable to a chest X-ray to diagnose pneumothorax or pneumomediastinum. Ultrasound in emergency may also be used to confirm a pneumothorax. Magnetic resonance imaging has no role in the acute investigation of DCI. One hundred per cent oxygen provides the maximum gradient for diffusion of nitrogen out of the bubbles. A large consecutive comparative series involving over 2000 divers has demonstrated that first aid oxygen significantly improves outcomes for divers with decompression illness. It should be administered in the pre-hospital setting and continue until and during recompression. Failure to improve on oxygen does not rule out DCI. Conversely, complete improvement on oxygen does not obviate the need for recompression. The diver should be supine or in the left lateral position if unable to protect their airway. Traditionally, the Trendelenburg position was advocated to reduce bubble embolization to the brain, but is now thought to increase the risk of cerebral oedema and should only be used if required to maintain blood pressure. The diver should be prevented from sitting or standing up, to avoid bubbles redistributing from the left ventricle to the brain. Initial resuscitation is along standard basic and advanced life support protocols. If intubation is required, the endotracheal tube cuff should be filled with saline prior to recompression to avoid a change in volume and a tube leak as ambient pressure increases. Intravenous isotonic crystalloids should be commenced and titrated to response. Glucose-containing fluids are to be avoided because they may exacerbate CNS injury. Divers who present after several days with mild symptoms may be adequately managed with oral fluids. A urinary catheter should be inserted for spinal cord DCI with bladder involvement. Hypothermia should be corrected. Long-distance retrieval can either be by air transport pressurized to 1 ATA or by portable recompression chambers. There is little debate that the longer the delay in recompression of severe DCI, the worse the outcome. However, Australian experience suggests that the number of cases where a portable chamber would have made a difference is so small that their use is unwarranted, largely because of the time required to prepare and transport portable chambers. Commercial aircraft are pressurized to 0.74 ATA (2440 m) and not appropriate to retrieve DCI patients, unless arrangements can be made to fly lower and pressurize to sea level. Road retrieval is not suitable over great distances or where an altitude of 300 m will be exceeded. Consultation with a hyperbaric physician should occur if retrieval is difficult. Recompression in a hyperbaric chamber is indicated even if the diver becomes asymptomatic with first aid, because otherwise many will relapse. The relapse may be more severe than the original presentation, due to the pathophysiological changes already initiated by bubbles in the microvasculature and tissues or redistribution of bubbles. Response to recompression is determined by time to recompression and the initial severity of injury. Recompression should always occur as soon as possible. It is particularly urgent for severe cases where treatment commenced later than 4 h after injury is associated with a poor response. Mild cases often respond despite longer delays to recompression. Two types of hyperbaric chamber are available to administer recompression treatment:
Environmental Emergencies
28.1 Heat-related illness
Introduction
Epidemiology and pathophysiology
Prevention
Clinical features
Exercise-associated collapse
Heatstroke
Serotonin toxicity
Neuroleptic malignant syndrome
Malignant hyperthermia
Clinical investigations
Treatment
Exercise-associated collapse
Heatstroke
Serotonin toxicity
Neuroleptic malignant syndrome
Malignant hyperthermia
Prognosis and disposition
28.2 Hypothermia
Introduction
Epidemiology and pathophysiology
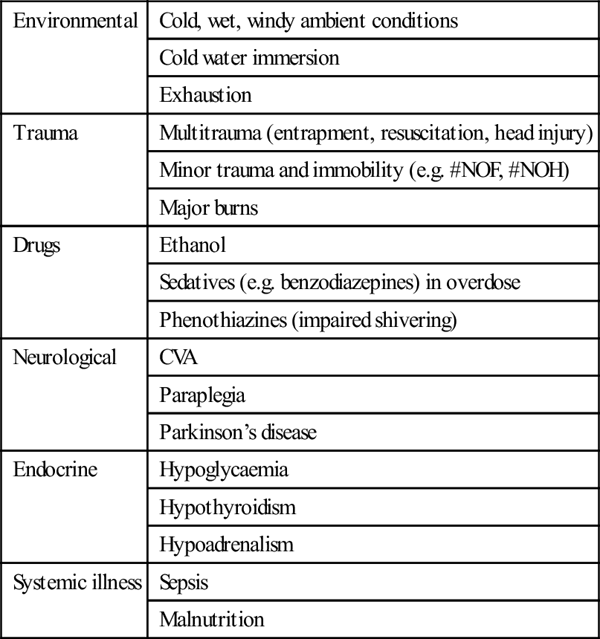
Environmental
Cold, wet, windy ambient conditions
Cold water immersion
Exhaustion
Trauma
Multitrauma (entrapment, resuscitation, head injury)
Minor trauma and immobility (e.g. #NOF, #NOH)
Major burns
Drugs
Ethanol
Sedatives (e.g. benzodiazepines) in overdose
Phenothiazines (impaired shivering)
Neurological
CVA
Paraplegia
Parkinson’s disease
Endocrine
Hypoglycaemia
Hypothyroidism
Hypoadrenalism
Systemic illness
Sepsis
Malnutrition
Clinical features
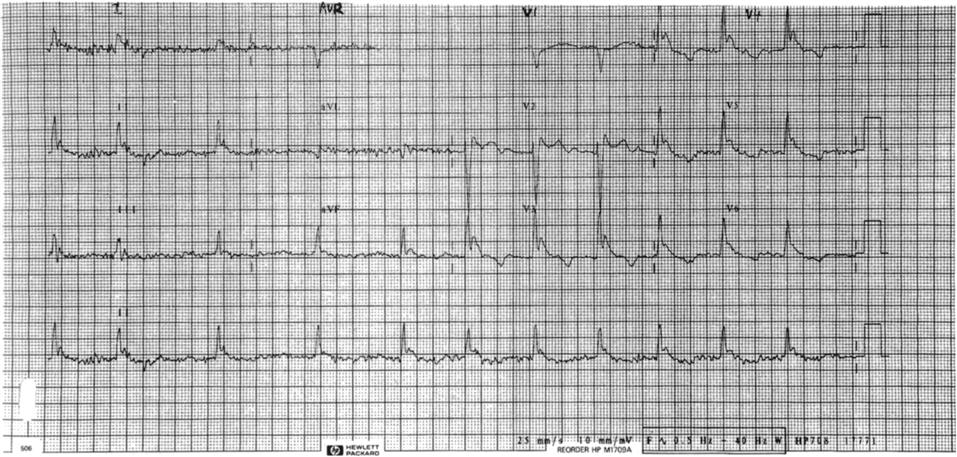
Clinical investigations
Treatment
General
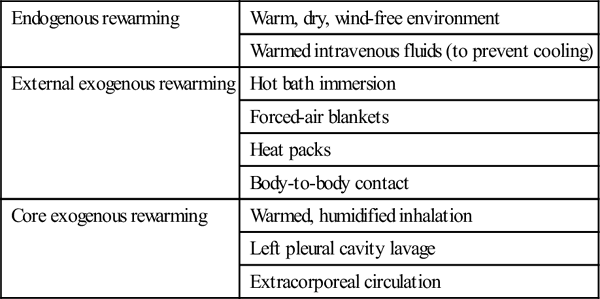
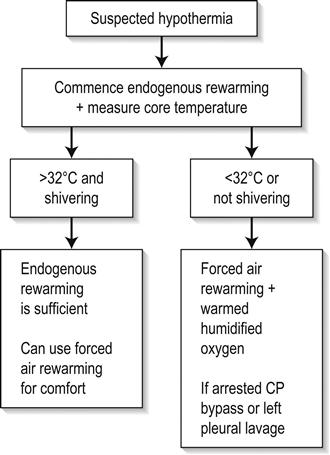
Rewarming therapies
Endogenous rewarming
Warm, dry, wind-free environment
Warmed intravenous fluids (to prevent cooling)
External exogenous rewarming
Hot bath immersion
Forced-air blankets
Heat packs
Body-to-body contact
Core exogenous rewarming
Warmed, humidified inhalation
Left pleural cavity lavage
Extracorporeal circulation
Prognosis and disposition
28.3 Dysbarism
Introduction
Diving physics and physiology
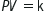
Depth (m)
Absolute pressure (ATA)
Gas volume (%)
0
1
100
10
2
50
20
3
33
30
4
25
40
5
20

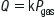
Barotrauma
Middle-ear barotrauma
Pathophysiology
Clinical features
Grade 0
Symptoms without signs
Grade 1
Injection of TM along handle of malleus
Grade 2
Slight haemorrhage within the TM
Grade 3
Gross haemorrhage within the TM
Grade 4
Free blood in middle ear
Grade 5
Perforation of TM
Treatment
Inner-ear barotrauma
Pathophysiology
Clinical features
Treatment
External ear barotrauma
Sinus barotrauma
Pathophysiology
Clinical features
Treatment
Mask squeeze
Gastrointestinal barotrauma
Dental barotrauma
Pulmonary barotrauma
Pathophysiology
Clinical features
Treatment
Decompression illness
Classification and criteria for diagnosis
 AGE can be caused by arterialization of venous bubbles released from tissues
AGE can be caused by arterialization of venous bubbles released from tissues
 Pre-hospital and emergency management prior to recompression is identical
Pre-hospital and emergency management prior to recompression is identical
 evolution of symptoms (spontaneously resolving/static/progressive/relapsing)
evolution of symptoms (spontaneously resolving/static/progressive/relapsing)
 body system affected (musculoskeletal/cutaneous/lymphatic/neurological/vestibular/cardiorespiratory)
body system affected (musculoskeletal/cutaneous/lymphatic/neurological/vestibular/cardiorespiratory)
Pathophysiology
Prevention
Clinical features
Assessment of the injured diver
Clinical investigations
Treatment
First aid
Retrieval
Recompression
Related posts:

Full access? Get Clinical Tree


28. Environmental Emergencies

Only gold members can continue reading. Log In or Register to continue
