Local anesthetics provide anesthesia and analgesia by blocking the transmission of pain sensation along nerve fibers.
 The key target of local anesthetics is the voltage-gated sodium channel. The binding is intracellular and is mediated by hydrophobic interactions.
The key target of local anesthetics is the voltage-gated sodium channel. The binding is intracellular and is mediated by hydrophobic interactions.
 The degree of nerve blockade depends on both drug concentration and volume.
The degree of nerve blockade depends on both drug concentration and volume.
 Most clinically relevant agents contain a lipid-soluble benzene ring connected to an amide group and are categorized as either aminoesters or aminoamides, based on their chemical linkage.
Most clinically relevant agents contain a lipid-soluble benzene ring connected to an amide group and are categorized as either aminoesters or aminoamides, based on their chemical linkage.
 Potency is related to hydrophobicity and physiochemical properties of the agent. In general, more potent agents are more lipid soluble.
Potency is related to hydrophobicity and physiochemical properties of the agent. In general, more potent agents are more lipid soluble.
 Efficacy for clinical use of local anesthetics may be increased by addition of epinephrine, opioids, and α2-adrenergic agonists. The value of alkalinization of local anesthetics appears to be debatable as a clinically useful tool to improve anesthesia.
Efficacy for clinical use of local anesthetics may be increased by addition of epinephrine, opioids, and α2-adrenergic agonists. The value of alkalinization of local anesthetics appears to be debatable as a clinically useful tool to improve anesthesia.
 The rate of local anesthetic system absorption depends on the site of injection, the dose, the drug’s intrinsic pharmacokinetic properties, and the addition of a vasoactive agent.
The rate of local anesthetic system absorption depends on the site of injection, the dose, the drug’s intrinsic pharmacokinetic properties, and the addition of a vasoactive agent.
 Systemic toxicity from the clinical use of local anesthetics is an uncommon occurrence. Patients with cardiovascular collapse from bupivacaine, ropivacaine, and levobupivacaine may be especially difficult to resuscitate; however, intravenous lipid infusion is a promising new therapy.
Systemic toxicity from the clinical use of local anesthetics is an uncommon occurrence. Patients with cardiovascular collapse from bupivacaine, ropivacaine, and levobupivacaine may be especially difficult to resuscitate; however, intravenous lipid infusion is a promising new therapy.
Multimedia
 Peripheral Nerve Cross-section
Peripheral Nerve Cross-section
 Ankle Block
Ankle Block
 Local anesthetics block the conduction of impulses in electrically excitable tissues. One of the important uses is to provide anesthesia and analgesia by blocking the transmission of pain sensation along nerve fibers. The molecular target of these agents is specific and the interaction has been extensively studied. Existing clinical applications are numerous and continue to expand. A comprehensive understanding of the mechanisms and the physiochemical properties of these agents would enable optimization of the therapeutic potential and avoid complications associated with inadvertent systemic toxicity.
Local anesthetics block the conduction of impulses in electrically excitable tissues. One of the important uses is to provide anesthesia and analgesia by blocking the transmission of pain sensation along nerve fibers. The molecular target of these agents is specific and the interaction has been extensively studied. Existing clinical applications are numerous and continue to expand. A comprehensive understanding of the mechanisms and the physiochemical properties of these agents would enable optimization of the therapeutic potential and avoid complications associated with inadvertent systemic toxicity.
MECHANISMS OF ACTION OF LOCAL ANESTHETICS
Anatomy of Nerves
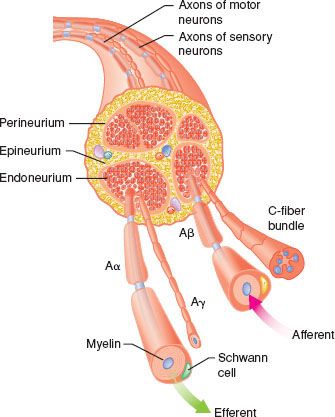
Local anesthetics are used to block nerves in the peripheral nervous system (PNS) and central nervous system (CNS). In the PNS, nerves contain both afferent and efferent fibers, which are bundled into one or more fascicles and organized within three tissue layers.1 Individual nerve fibers within each fascicle are surrounded by the endoneurium, a loose connective tissue containing glial cells, fibroblasts, and blood capillaries. A dense layer of collagenous connective tissue called the perineurium surrounds each fascicle. A final layer of dense connective tissue, the epineurium, encases groups of fascicles into a cylindrical sheath (Fig. 21-1). These layers of tissue offer protection to the surrounded nerve fibers and act as barriers to passive diffusion of local anesthetics.2
FIGURE 21-1. Schematic cross section of typical peripheral nerve. The epineurium, consisting of collagen fibers, is oriented along the long axis of the nerve. The perineurium is a discrete cell layer, whereas the endoneurium is a matrix of connective tissue. Both afferent and efferent axons are shown. Sympathetic axons (not shown) are also present in mixed peripheral nerves. (Adapted from: Strichartz GR. Neural physiology and local anesthetic action. In: Cousins MJ, Bridenbaugh PO, eds. Neural Blockade in Clinical Anesthesia and Management of Pain. Philadelphia, PA: Lippincott-Raven; 1998:35, with permission.)
Nerves in both the CNS and PNS are differentiated by the presence or absence of myelin sheath. Myelinated nerve fibers are surrounded by Schwann cells in the PNS and by oligodendrocytes in the CNS. The cells form a concentrically wrapped lipid bilayer sheath around the axons that cover the length of the nerve.3 The myelin sheath is interrupted at short, regular intervals by specialized regions called nodes of Ranvier, which contain densely clustered protein elements essential for transmission of neuronal signals4 (Fig. 21-2). As electrical signals are renewed at each node, nerve impulses move in myelinated fibers by saltatory conduction. In contrast, there are no nodes of Ranvier in nonmyelinated nerve fibers. Although these nerve fibers are similarly encased in Schwann cells, the plasma membrane does not wrap around the axons concentrically. Several nerve fibers may be simultaneously embedded within a single Schwann cell1 (Fig. 21-3).
FIGURE 21-3. Schwann cells form myelin around one myelinated axon or encompass several unmyelinated axons. (Adapted from: Carpenter RL, Mackey DC. Local anesthetics. In: Barash PG, Cullen BF, Stoelting RF, eds. Clinical Anesthesia. 3rd ed. Philadelphia, PA: Lippincott-Raven; 1996:413, with permission.)
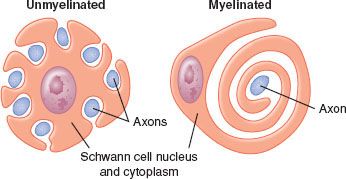
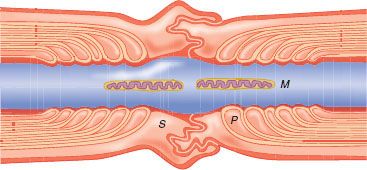
FIGURE 21-2. Diagram of node of Ranvier displaying mitochondria (M), tight junctions in paranodal area (P), and Schwann cell (S) surrounding node. (Adapted from: Strichartz GR. Mechanisms of action of local anesthetic agents. In: Rogers MC, Tinker JH, Covino BG, et al., eds. Principles and Practice of Anesthesiology. St. Louis, MO: Mosby Year Book; 1993:1197, with permission.)
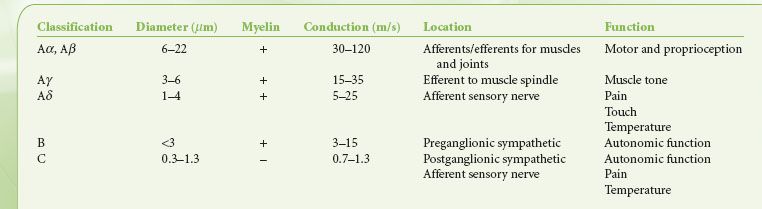
Nerve fibers are commonly classified according to their size, conduction velocity, and function (Table 21-1). In general, nerve fibers with cross-sectional diameter greater than 1 μm are myelinated. Both a larger nerve size and the presence of myelin sheath are associated with faster conduction velocity.5 Nerve fibers with large diameters have better intrinsic electric conductance. Myelin improves the electrical insulation of nerve fibers and permits more rapid impulse transmission via saltatory conduction. Large-diameter, myelinated fibers, many of which are classified as A fibers, are typically involved in motor and sensory functions in which speed of nerve transmission is critical. In contrast, small-diameter, nonmyelinated C fibers have slower conduction velocity and relay sensory information such as pain, temperature, and autonomic functions.
TABLE 21-1. CLASSIFICATION OF NERVE FIBERS
Electrophysiology of Neural Conduction and Voltage-gated Sodium Channels
Transmission of electrical impulses along the cell membrane forms the basis of signal transduction along nerve fibers. Energy necessary for the propagation and maintenance of the electric potential is maintained on the cell surface by ionic disequilibria across the semipermeable cell membrane.6 The resting membrane potential, approximately –60 to –70 mV in neurons (the extracellular electric potential is, by convention, defined as zero, and the intracellular potential is thus negative relative to it), is derived predominantly from a difference in the intracellular and extracellular concentrations of potassium and sodium ions. Neurons at rest are more permeable to potassium ions than sodium ions because of potassium leak channels; therefore membrane potential is closer to the equilibrium potential of potassium (EK –80 mV) than that of sodium (ENa +60 mV). The ion gradient is continuously regenerated by protein pumps, cotransporters, and channels via adenosine triphosphate–dependent process.
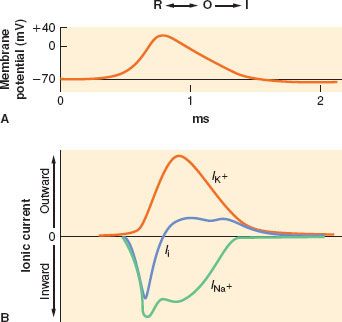
Electrical impulses are conducted along nerve fibers as action potentials. They are brief, localized spikes of positive charge, or depolarizations, on the cell membrane caused by rapid influx of sodium ions down its electrochemical gradient.7 An action potential is initiated by local membrane depolarization, such as at the cell body or nerve terminal by ligand–receptor complex. When a certain charge threshold is reached, an action potential is triggered and further depolarization occurs in an “all-or-none” fashion.8 The spike in membrane potential peaks around +50 mV, at which point the influx of sodium is replaced with an efflux of potassium, causing a reversal of membrane potential, or repolarization. The passive diffusion of membrane depolarization triggers other action potentials in either adjacent cell membrane in nonmyelinated nerve fibers or adjacent nodes of Ranvier in myelinated nerve fibers, resulting in a wave of action potential being propagated along the nerve. A short refractory period that ensues after each action potential prevents the retrograde spread of action potential on previously activated membranes.7
The flow of ions responsible for action potentials is mediated by a variety of channels and pumps, the most important of which are the voltage-gated sodium channels. They are essential for the influx of sodium ions during the rapid depolarization phase of the action potential and belong to a family of channel proteins that also includes voltage-gated potassium and voltage-gated calcium channels. Each voltage-gated sodium channel is a complex made up of one principal α-subunit and one or more auxiliary β-subunits.9 The α-subunit is a single-polypeptide transmembrane protein that contains most of the key components of the channel function. They include four homologous α-helical domains (D1 to D4) that form the channel pore and control ion selectivity, voltage-sensing regions that regulate gating function and inactivation, and phosphorylation sites for modulation by protein kinases. β-subunits are short polypeptide proteins with a single transmembrane domain. They are linked to α-subunits by either noncovalent or disulfide bonds; although they are dispensable for channel activity, evidence suggests that they perhaps play a role in modulation of channel expression, localization, and function.
In the absence of a stimulus, voltage-gated sodium channels exist predominantly in the resting or closed state (Fig. 21-4). On membrane depolarization, positive charges on the membrane interact with charged amino acid residues in the voltage-sensing regions (S4).10 This induces a conformational change in the channel, converting it to the open state. Sodium ions rush through the opened pore, which is lined with negatively charged residues. Ion selectivity is determined by these amino acid residues; changes in their composition can lead to increased permeability for other cations, such as potassium and calcium.11 Within milliseconds after opening, channels undergo a transition to the inactivated state. Depending on the frequency and voltage of the initial depolarizing stimulus, the channel may undergo either fast or slow inactivation. Slow or fast inactivation refers to the duration in which the channel remains refractory to repeat depolarization before resetting to the closed state. Fast inactivation completes within a millisecond and is sensitive to the action of local anesthetics. It is mediated by a short mobile intracellular polypeptide loop connecting domains D3 and D4 that closes the channel from inside the cell via a hinge-lid mechanism.12 A triad of highly hydrophobic amino acids (isoleucine, phenylalanine, and methionine [IFM]) appears to be an important structural determinant of fast activation; disrupting the loop or changing the hydrophobicity of the amino acids abrogates fast inactivation.13,14 Slow activation, lasting seconds to minutes, is distinct from fast activation. It is resistant to the action of local anesthetics and its mechanism is less well understood. It often occurs after prolonged depolarization and is believed to be important in regulating membrane excitability.
FIGURE 21-4. Illustration of dominant form of sodium channel during generation of an action potential. R, resting form; O, open form; I, inactive form. (A) The concurrent generation of an action potential as the membrane depolarizes from resting potential. (B) The concurrent changes in ion flux, as inward sodium current (INA+) and outward potassium current (IK+) together yield the net ionic current across the membrane (Ii). (Adapted from: Strichartz GR. Neural physiology and local anesthetic action. In: Cousins MJ, Bridenbaugh PO, eds. Neural Blockade in Clinical Anesthesia and Management of Pain. Philadelphia, PA: Lippincott-Raven; 1998:35, with permission.)
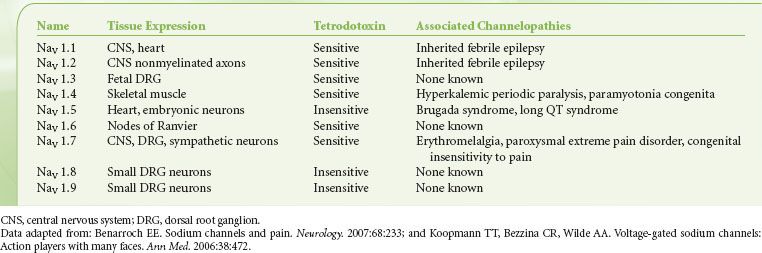
Nine isoforms of voltage-gated sodium channels (NaV 1.1 to NaV 1.9) have been identified; each relates to a unique α-subunit subtype (Table 21-2). Each isoform varies slightly in its channel kinetics, such as threshold of activation and mode of inactivation, and its sensitivity to blocking agents like tetrodotoxins and local anesthetics. Cell and tissue expression of individual isoforms may be quite specific; for instance, NaV 1.2 is found almost exclusively in the CNS, whereas NaV 1.6 is restricted to nodes of Ranvier in both CNS and PNS.15 Likewise, several isoforms could be present on a single cell type; both NaV 1.8 and NaV 1.9 have been found in small- to medium-sized neurons in dorsal root ganglions that are connected to Aδ and C fibers. Whether individual isoforms each have a separate and defined role remains to be seen; however, clues to their function may be inferred from studies of several inherited diseases that have been associated with sodium channelopathies. Hyperexcitability of NaV 1.7 has been implicated in several painful disease states, such as primary erythromelalgia and paroxysmal extreme pain disorder.16,17 Conversely, null mutation of NaV 1.7 is linked to a rare genetic condition in which otherwise normal individuals have severely impaired perception to pain.18,19
TABLE 21-2. VOLTAGE-GATED SODIUM CHANNELS
Molecular Mechanisms of Local Anesthetics
 Local anesthetics block the transmission of nerve impulses by targeting the function of voltage-gated sodium channels. Although several local anesthetics can bind to other receptors like voltage-gated potassium channels and nicotinic acetylcholine receptors and their amphipathic nature may enable them to interact with plasma membranes, it is widely accepted that they induce anesthesia and analgesia through direct interactions with the sodium channels. Other molecules with local anesthetic properties such as tricyclic antidepressants and anticonvulsants may likewise interact with voltage-gated sodium channels; however, it is unclear if they act through similar mechanisms. Therefore, the following discussion is limited to the “traditional” set of local anesthetic molecules.
Local anesthetics block the transmission of nerve impulses by targeting the function of voltage-gated sodium channels. Although several local anesthetics can bind to other receptors like voltage-gated potassium channels and nicotinic acetylcholine receptors and their amphipathic nature may enable them to interact with plasma membranes, it is widely accepted that they induce anesthesia and analgesia through direct interactions with the sodium channels. Other molecules with local anesthetic properties such as tricyclic antidepressants and anticonvulsants may likewise interact with voltage-gated sodium channels; however, it is unclear if they act through similar mechanisms. Therefore, the following discussion is limited to the “traditional” set of local anesthetic molecules.
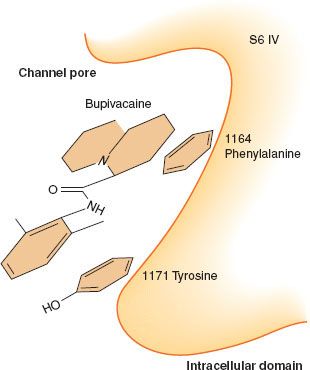
Local anesthetics reversibly bind the intracellular portion of voltage-gated sodium channels (Fig. 21-5). Early experiments with giant squid axons demonstrated that a derivative of lidocaine with a permanent positive charge, QX314, which cannot cross the plasma membrane, blocks ion current through voltage-gated sodium channels only with intra-axoplasmic injections, but not with external application.20 Subsequent mutational analyses have supported the observation and identified specific sites on the channel involved in drug recognition.21 Several hydrophobic aromatic residues (a phenylalanine at position 1,764 and a tyrosine at position 1,771 in NaV 1.2) located within an α-helix (S6) of domains 1, 3, and 4 are essential for drug binding (Fig. 21-6). They line an inner cavity within the intracellular portion of the channel pore and span a region about 11 Å apart, roughly the size of a local anesthetic molecule. Changes in either residue severely reduce the binding affinity. Another hydrophobic amino acid (an isoleucine at position 1,760), located near the outer pore opening, has similarly been found to influence the dissociation of local anesthetics from the channel by antagonizing the release of drugs through the channel pore.
FIGURE 21-5. Diagram of bilayer lipid membrane of conductive tissue with sodium channel spanning the membrane. Tertiary amine local anesthetics exist as neutral base (N) and protonated, charged form (NH+) in equilibrium. The neutral base (N) is more lipid soluble, preferentially partitions into the lipophilic membrane interior, and easily passes through the membrane. The charged form (NH+) is more water soluble and binds to the sodium channel at the negatively charged membrane surface. Both forms can affect the function of the sodium channel. The N form can cause membrane expansion and closure of the sodium channel. The NH+ form will directly inhibit the sodium channel by binding with a local anesthetic receptor. The natural “local anesthetic” tetrodotoxin (TTX) binds at the external surface of the sodium channel and has no interaction with the clinically used local anesthetics. (Adapted from: Strichartz GR. Neural physiology and local anesthetic action. In: Cousins MJ, Bridenbaugh PO, eds. Neural Blockade in Clinical Anesthesia and Management of Pain. Philadelphia, PA: Lippincott-Raven; 1998:35, with permission.)
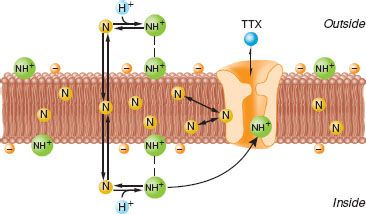
FIGURE 21-6. Diagram of local anesthetic–binding site, depicting a hydrophobic pocket within the channel pore. (Adapted from: Ragsdale DS, McPhee JC, Scheuer T, et al. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724, with permission.)
Application of local anesthetics typically produces a concentration-dependent decrease in the peak sodium current.22,23 Known as “tonic blockade,” it reflects the reduction in the number of sodium channels for a given drug concentration present in the open state at equilibrium. In contrast, repetitive stimulation of the sodium channels often leads to a shift in the steady-state equilibrium, resulting in a greater number of channels being blocked at the same drug concentration. Termed use-dependent blockade, the exact mechanism is incompletely understood and has been the subject of many competing hypotheses. One popular theory, the modulated-receptor theory, proposes that local anesthetics bind to the open or the inactivated channels more avidly than the resting channels, suggesting that drug affinity is a function of a channel’s conformational state. An alternate theory, the guarded-receptor theory, assumes that the intrinsic binding affinity remains essentially constant regardless of a channel’s conformation; rather, the apparent affinity is associated with increased access to the recognition site resulting from channel gating. Experimental evidence so far has been inconclusive.
Mechanism of Nerve Blockade
Local anesthetics block peripheral nerves by disrupting the transmission of action potentials along nerve fibers. To get to its site of action, principally the voltage-gated sodium channels, local anesthetics have to reach the targeted nerve membrane. This entails the diffusion of drugs through tissues and the generation of a concentration gradient. Even with close proximity of deposition, only about 1% to 2% of injected local anesthetics ultimately penetrate into the nerve.24 As discussed earlier, the perineural sheath encasing nerve fibers appears to be an important determinant; nerves that have been desheathed in vitro require about a hundred-fold lower local anesthetic concentration (in the 0.7 to 0.9 mM range for lidocaine) than nerves in vivo (the typical 2% lidocaine used clinically is equivalent to 75 mM concentration). Although it may vary with anatomic location and nerve physiology, functional block typically occurs within 5 minutes of injection in rat sciatic nerves, and this time course corresponds to the peak in the intraneural drug absorption.
 The degree of nerve blockade depends on the local anesthetic concentration and volume. For a given drug, a minimal concentration is necessary to effect complete nerve blockade. It reflects the potency of the local anesthetics and the intrinsic conduction properties of nerve fibers, which in turn likely depend on the drug’s binding affinity to the ion channels and the degree of drug saturation necessary to halt the transmission of action potentials. Accordingly, individual types of nerve fibers differ in their minimal blocking concentration, such that some A fibers are blocked by lower drug concentrations than C fibers.25 Likewise, the pattern of stimulation (tonic vs. use-dependent blockade) influences the degree of conduction failure; repetitive stimulations, which can lead to a shift in steady-state equilibrium of blocked sodium channels, are associated with higher conduction failure than tonic stimulation at a given drug concentration.26
The degree of nerve blockade depends on the local anesthetic concentration and volume. For a given drug, a minimal concentration is necessary to effect complete nerve blockade. It reflects the potency of the local anesthetics and the intrinsic conduction properties of nerve fibers, which in turn likely depend on the drug’s binding affinity to the ion channels and the degree of drug saturation necessary to halt the transmission of action potentials. Accordingly, individual types of nerve fibers differ in their minimal blocking concentration, such that some A fibers are blocked by lower drug concentrations than C fibers.25 Likewise, the pattern of stimulation (tonic vs. use-dependent blockade) influences the degree of conduction failure; repetitive stimulations, which can lead to a shift in steady-state equilibrium of blocked sodium channels, are associated with higher conduction failure than tonic stimulation at a given drug concentration.26
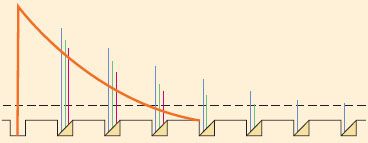
Of equal importance as drug concentration is the local anesthetic volume. A sufficient volume is needed to suppress the regeneration of nerve impulse over a critical length of nerve fiber. According to the model of decremental conduction (Fig. 21-7), as membrane depolarization from an action potential passively decays with distance along nerve fibers, the presence of local anesthetics decreases the ability of adjacent membrane or successive nodes of Ranvier to regenerate the impulse.27 Transmission stops once the membrane depolarization falls below the threshold for action potential activation. Too short an exposure length allows impulses to “skip” over membranes or nodes that are blocked by even the highest drug concentration, whereas exposure of a long nerve segment to a relatively low drug concentration can still result in gradual extinction of impulses by decremental decay.
FIGURE 21-7. Diagram illustrating the principle of decremental conduction block by local anesthetic at a myelinated axon. The first node of Ranvier at left contains no local anesthetic and gives rise to a normal action potential (solid curve). If the nodes succeeding the first are occupied by a concentration of local anesthetic high enough to block 74% to 84% of the sodium conductance, then the action potential amplitudes decrease at successive nodes (amplitudes are indicated by interrupted bars representing three increasing concentrations of local anesthetic). Eventually, the impulse decays to below-threshold amplitude if the series of local anesthetic–containing nodes is long enough. Propagation of the impulse is then blocked by decremental conduction, even though none of the nodes are completely blocked. Concentrations of local anesthetics that block more than 84% of the sodium conductance at three successive nodes prevent any impulse propagation at all. (Adapted from: Fink BR. Mechanisms of differential axial blockade in epidural and spinal anesthesia. Anesthesiology. 1989;70:851, with permission.)
Not all sensory and motor modalities are blocked equally by local anesthetics. It has long been observed that application of local anesthetics produced an ordered progression of sensory and motor deficits, starting commonly with the disappearance of temperature sensation, followed by proprioception, motor function, sharp pain, and then light touch. Termed differential blockade, historically this had been thought to be related simply to the diameter of the nerve fibers, with the smaller fibers inherently more susceptible to drug blockade than larger fibers.28 However, while the “size principle” of differential blockade is consistent with many experimental findings, it does not appear to be universally true. Larger, myelinated Aδ fibers (believed to mediate sharp pain) have been found to be blocked preferentially over small, nonmyelinated C fibers (dull pain). Furthermore, within the C fibers are fast and slow components of impulse transmission, each with distinct susceptibility to drug blockade.29 These observations argue against a purely pharmacokinetic mechanism as the sole basis for explaining differential blockade. Instead, it may also likely depend on the intrinsic excitatory properties of the nerve fibers, namely, the patterned expression of voltage-gated sodium channels. Indeed, two channel isoforms, NaV 1.7 and NaV 1.8, both present on dorsal root ganglions, have been shown to possess different sensitivities to lidocaine blockade.30 It remains to be seen how differential blockade of certain channels might translate into selective inhibition of pain and other sensory modalities.
PHARMACOLOGY AND PHARMACODYNAMICS
Chemical Properties and Relationship to Activity and Potency
 Most clinically relevant local anesthetics are made up of a lipid-soluble, aromatic benzene ring connected to an amide group via either an amide or an ester moiety. The type of linkage divides them broadly into two categories, the aminoesters and the aminoamides. Aside from a difference in their metabolic pathways (aminoesters are hydrolyzed by plasma cholinesterases and aminoamides are degraded by hepatic carboxylesterase) and the incidence of allergic reactions attributed, their membership in either category offers little in distinction of their biophysical properties. Rather, the distinguishing physiochemical characteristics are associated with the alkalinity of the amide group, the lipophilicity conferred by the alkyl substitution on the amide group and the benzene ring, and the stereochemistry of related isomers.
Most clinically relevant local anesthetics are made up of a lipid-soluble, aromatic benzene ring connected to an amide group via either an amide or an ester moiety. The type of linkage divides them broadly into two categories, the aminoesters and the aminoamides. Aside from a difference in their metabolic pathways (aminoesters are hydrolyzed by plasma cholinesterases and aminoamides are degraded by hepatic carboxylesterase) and the incidence of allergic reactions attributed, their membership in either category offers little in distinction of their biophysical properties. Rather, the distinguishing physiochemical characteristics are associated with the alkalinity of the amide group, the lipophilicity conferred by the alkyl substitution on the amide group and the benzene ring, and the stereochemistry of related isomers.
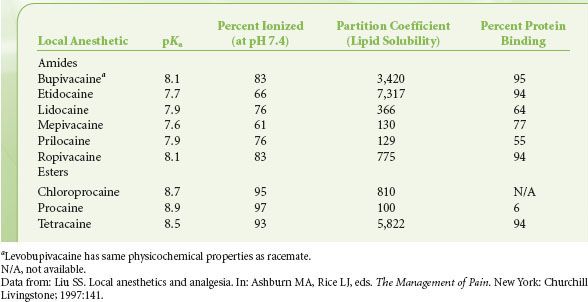
The tertiary amide found in local anesthetics is capable of accepting a proton, albeit with low affinity; thus, these compounds are classified as weak bases. At physiologic pH, local anesthetics in solution are in equilibrium between the protonated, cationic form and the lipid-soluble, neutral forms. The ratio of the two forms depends on the pKa or the dissociation constant of the local anesthetics and the surrounding pH (Table 21-3). A ratio with high concentration of the lipid-soluble form favors entry into the cell, as the main pathway for entry is by passive adsorption of lipid-soluble form through the cell membrane.2 Clinically, alkalization of the anesthetic solution increases the ratio of the lipid-soluble form to cationic form, thereby facilitating cell entry. Once inside the cell, equilibrium is reestablished between the cationic and the neutral forms, and it appears that the cationic form is more potent of the two in its activity on sodium channels.31
TABLE 21-3. PHYSICOCHEMICAL PROPERTIES OF CLINICALLY USED LOCAL ANESTHETICS
Lipid solubility of local anesthetics is determined by the degree of alkyl group substitution on the amide group and the benzene ring. In the laboratory, it is measured by the partition coefficient in octanol, a hydrophobic solvent, and compounds with high octanol:buffer partition coefficients are more lipid soluble.32 A positive correlation exists between the potency of the local anesthetics and their octanol:buffer partition coefficient; highly lipid-soluble agents are more potent and tend to have a longer duration of action than ones that are less lipid soluble.33 This is consistent with experimental findings that show local anesthetics bind to a hydrophobic pocket within the sodium channels, suggesting that ligand binding may be mediated primarily by hydrophobic and van der Waals interactions21 (Fig. 21-6).
 Whereas the correlation between local anesthetic potency and hydrophobicity generally holds true in vitro, it may not be as exact in vivo. As opposed to setups with isolated nerves, other factors may influence the potency of local anesthetics on nerves in situ.34 For example, highly lipid-soluble agents may be sequestered into surrounding adipose cells. Vasodilatory properties of local anesthetics may likewise alter drug redistribution into the neighboring tissues.35,36 Relative potency of local anesthetics has been determined for different clinical applications and these values are listed in Table 21-4.
Whereas the correlation between local anesthetic potency and hydrophobicity generally holds true in vitro, it may not be as exact in vivo. As opposed to setups with isolated nerves, other factors may influence the potency of local anesthetics on nerves in situ.34 For example, highly lipid-soluble agents may be sequestered into surrounding adipose cells. Vasodilatory properties of local anesthetics may likewise alter drug redistribution into the neighboring tissues.35,36 Relative potency of local anesthetics has been determined for different clinical applications and these values are listed in Table 21-4.
TABLE 21-4. RELATIVE POTENCY OF LOCAL ANESTHETICS FOR DIFFERENT CLINICAL APPLICATIONS

Finally, anesthetic activity and potency are affected by the stereochemistry of local anesthetics. Many older drugs exist as racemic mixtures; that is, enantiomeric stereoisomers differing in the arrangement at the asymmetric or chiral carbon atom are in equal proportion. Newer agents, namely, ropivacaine and levobupivacaine, are available as purely single enantiomers. They were initially developed as less cardiotoxic alternatives to bupivacaine. While the desired improvement in the safety index has been generally supported by clinical studies, it appears that, overall, this is at the expense of a slight decrease in potency and shorter duration of action compared with the racemic mixtures.37,38 A theoretical basis for the difference between the enantiomeric stereoisomers can be readily hypothesized; however, little is known of the exact mechanisms involved. Likely, it may include subtle stereoselective preference in local anesthetic binding among the individual sodium channel isoforms.
Additives to Increase Local Anesthetic Activity
 Epinephrine
Epinephrine
Reported benefits of epinephrine include prolongation of local anesthetic block, increased intensity of block, and decreased systemic absorption of local anesthetic.39 Epinephrine’s vasoconstrictive effects augment local anesthetics by antagonizing inherent vasodilating effects of local anesthetics, decreasing systemic absorption and intraneural clearance, and perhaps by redistributing intraneural local anesthetic.39,40
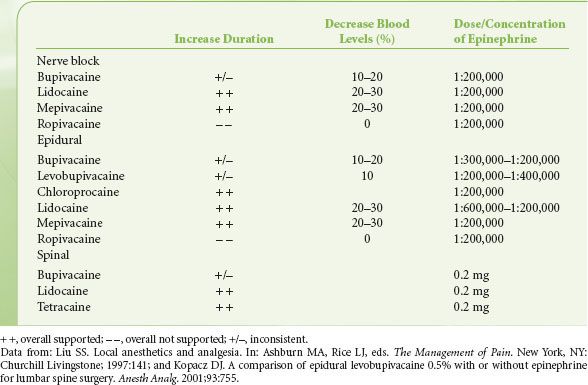
Direct analgesic effects from epinephrine may also occur via interaction with α2-adrenergic receptors in the brain and spinal cord,41 especially because local anesthetics increase the vascular uptake of epinephrine.42 The clinical effects of the use of epinephrine are listed in Table 21-5. The smallest dose is suggested because epinephrine combined with local anesthetics may have toxic effects on tissue,43 the cardiovascular system,44 peripheral nerves, and the spinal cord.39
TABLE 21-5. EFFECTS OF ADDITION OF EPINEPHRINE TO LOCAL ANESTHETICS
Alkalinization of Local Anesthetic Solution
Local anesthetic solutions are alkalinized in order to hasten onset of neural block.45 The pH of commercial preparations of local anesthetics ranges from 3.9 to 6.47 and is especially acidic if prepackaged with epinephrine.46 As the pKa of commonly used local anesthetics ranges from 7.6 to 8.9 (Table 21-3), less than 3% of the commercially prepared local anesthetic exists as the lipid-soluble neutral form. As previously discussed, the neutral form is believed to be important for penetration into the neural cytoplasm, whereas the charged form primarily interacts with the local anesthetic receptor within the sodium channel. Therefore, the rationale for alkalinization was to increase the ratio of local anesthetic existing as the lipid-soluble neutral form. However, clinically used local anesthetics cannot be alkalinized beyond a pH of 6.05 to 8 before precipitation occurs46 and these pH values will only increase the neutral form to about 10%.
Clinical studies on the association between alkalinization of local anesthetics and hastening of block onset have shown an improvement of less than 5 minutes compared with commercial preparations.45,47 Furthermore, results from a study in rats indicate that alkalinization of lidocaine may also decrease the duration of peripheral nerve blocks if the mixture contained no epinephrine.48 Together, alkalinization of local anesthetics appears limited as a clinically useful adjuvant to improving anesthesia.
Opioids
Opioids have multiple central and peripheral mechanisms of analgesic action (see Chapter 19: Opioids). Spinal administration of opioids provides analgesia primarily by attenuating C-fiber nociception49 and is independent of supraspinal mechanisms.50 Coadministration of opioids with central neuraxial local anesthetics results in synergistic analgesia.51 An exception to this analgesic synergy is chloroprocaine, which appears to decrease the effectiveness of opioids coadministered epidurally.52 The reason is unclear but the mechanism does not seem to involve direct antagonism of opioid receptors.53 Nonetheless, clinical studies support the practice of central neuraxial coadministration of local anesthetics and opioids for prolongation and intensification of analgesia and anesthesia.51
The discovery of peripheral opioid receptors initially generated much interest in the use of opioids as adjuvants to local anesthetics for peripheral nerve blockade.54 However, while some studies have reported favorable outcome of such coadministration, others have failed to demonstrate any increased efficacy.55 A problem that has plagued many studies is the lack of adequate controls for differentiating the analgesic effects of opioids acting peripherally versus a more central mechanism resulting from systemically absorbed opioids. Nonetheless, recent carefully designed trials have shown that some opioids, namely, buprenorphine, may enhance and prolong postoperative analgesia better than either local anesthetics alone or local anesthetics administered with intramuscular buprenorphine.56,57 Finally, cumulative evidence does not support the use of intraarticular coadministration of local anesthetic and opioid for postoperative analgesia.58
α2-adrenergic Agonists
α2-adrenergic agonists can be a useful adjuvant to local anesthetics. α2-specific agonists such as clonidine produce analgesia via supraspinal and spinal adrenergic receptors.59 Clonidine also has direct inhibitory effects on peripheral nerve conduction (A and C nerve fibers).60 Thus, addition of clonidine may have multiple routes of action depending on the type of application. Preliminary evidence suggests that coadministration of an α2-agonist and local anesthetic results in central neuraxial and peripheral nerve analgesic synergy,61 whereas systemic (supraspinal) effects are additive.62 On average, clonidine improves the duration of analgesia by about 2 hours, regardless of whether an intermediate- or long-acting local anesthetic is used.63 Overall, results from clinical trials indicate that clonidine can enhance local anesthetics when used for intrathecal and epidural anesthesia and peripheral nerve blocks.63,64
Steroids
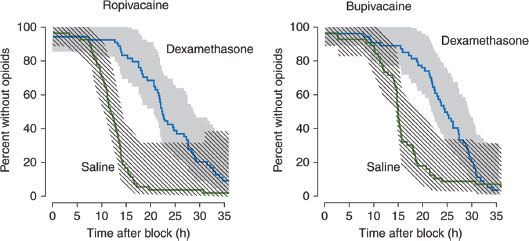
Potent glucocorticoid injections have been widely used for the treatment of chronic low back pain caused by radiculopathy. Experiments in animals using extended-release preparations of local anesthetics have found that addition of dexamethasone to the mixture prolongs the conduction block after peripheral nerve application.65,66 The duration of the blockade is associated with the potency of the glucocorticoid activity and appears to be steroid receptor dependent and locally mediated.67 Clinical reports of the use of dexamethasone as an adjuvant to local anesthetics have shown similar prolongation of anesthesia after brachial plexus blockades68,69 and intravenous regional anesthesia.70 Combined with intermediate- to long-acting local anesthetics, dexamethasone extends the duration of analgesia by approximately 50% after utilization of the supraclavicular68 or interscalene approach69 to the brachial plexus block (Fig. 21-8). Although initial laboratory data show no evidence of increased neurotoxicity from use of dexamethasone as compared with other adjuvants, our current understanding of its mechanism of action and potential side effects remains incomplete.71
FIGURE 21-8. Addition of dexamethasone to either ropivacaine or bupivacaine increases the duration of analgesia after interscalene brachial plexus block. Data shown as the Kaplan–Meier survival density estimates with the shaded region representing the 95% confidence interval. (Reprinted from: Cummings KC, Napierkowski DE, Parra-Sanchez I, et al. Effect of dexamethasone on the duration of interscalene nerve blocks with ropivacaine or bupivacaine. Br J Anaesth. 2011;107:446, with permission.)






Full access? Get Clinical Tree


