Drugs like antineoplastic agents can produce thrombocytopenia by suppressing platelet production in the bone marrow, but the most common mechanism for drug-induced thrombocytopenia is the production of antibodies that cross-react with platelets (8). This immune-mediated thrombocytopenia is most frequently observed with heparin, and less frequently with platelet glycoprotein receptor (IIb/IIIa) antagonists and selected antibiotics (particularly linezolid, β-lactams, and vancomycin).
Heparin-Induced Thrombocytopenia
There are two types of thrombocytopenia associated with heparin. The first is a nonimmune response that results in mild thrombocytopenia (platelet counts may not fall below 100,000/∝L) in the first few days after starting heparin. This reaction is reported in 10 to 30% of patients receiving heparin (10), and it resolves spontaneously without interruption of heparin, and without adverse consequences. The second type of thrombocytopenia is an immune response that typically appears 5 to 10 days after starting heparin (10,11). This reaction is much less common (incidence = 1–3%) but is much more serious; i.e., it can produce life-threatening thrombosis (not hemorrhage), and has a mortality rate as high as 30% if left unnoticed (10). The term heparin-induced thrombocytopenia (HIT) is reserved for the immune-mediated thrombocytopenia, and this condition is the focus of the current presentation.
Pathogenesis
Heparin is not immunogenic itself, but it binds to a protein (platelet factor 4) on platelets to form an antigenic complex that can trigger the formation of IgG antibodies. These antibodies bind to platelets and induce a strong platelet activation response to promote thrombosis. These antibodies can also bind to endothelial cells and promote the release of tissue factor from the endothelium; this promotes fibrin formation and further accelerates the thrombotic process. The reticuloendothelial system can clear antibody-coated platelets, and this helps to limit the incidence of thrombosis. Heparin-associated antibodies usually disappear within 3 months after discontinuing heparin (10).
Risk Factors
One of the most important features of HIT is the fact that it is not a dose-dependent reaction, and can occur as a result of heparin exposure from heparin-based flushes of intravascular catheters, or even heparin-coated pulmonary artery catheters (12). The type of heparin preparation does, however, influence the risk of HIT; i.e., the risk of HIT is ten times greater with unfractionated heparin (UFH) than with low-molecular-weight heparin (LMWH) (11). The risk of HIT also varies by patient population; i.e., it is highest in patients undergoing orthopedic and cardiac surgical procedures, and lowest in medical patients (10,11). The reported incidence of HIT with UFH is 1–5% following orthopedic or cardiac surgery, and 0.1–1% in medical patients (11).
Clinical Features
HIT typically appears 5 to 10 days after the first exposure to heparin, but can appear within 24 hours in patients with HIT antibodies due to heparin exposure within the past 3 months (11). Platelet counts are usually between 50,000/∝L and 150,000/∝L. Severe thrombocytopenia (<20,000/mL) is uncommon in HIT (10,11). In up to 25% of cases of HIT, the thrombosis precedes the thrombocytopenia (11).
THROMBOSIS: Venous thrombosis is more common than arterial thrombosis. Reports indicate that 17% to 55% of patients with untreated HIT develop deep vein thrombosis in the legs and/or pulmonary embolism, whereas only 1% to 3% of patients develop arterial thromboses resulting in limb ischemia, thrombotic stroke, or acute myocardial infarction (11). Limb gangrene from thrombotic veno-occlusion has been reported in 5% to 10% of patients with HIT who are treated with a vitamin K antagonist (e.g., coumadin).
Diagnosis
About 8 different assays are currently used to detect HIT antibodies. The most popular of these is an enzyme-linked immunosorbent assay (ELISA) for antibodies to the platelet factor 4-heparin complex. A negative assays helps to exclude the diagnosis of HIT, but a positive assay does not confirm the diagnosis because HIT antibodies do not always promote thrombocytopenia or thrombosis (11). The diagnosis of HIT requires a positive antibody assay in combination with a high index of clinical suspicion.
Table 19.2 Anticoagulation with Direct Thrombin Inhibitors
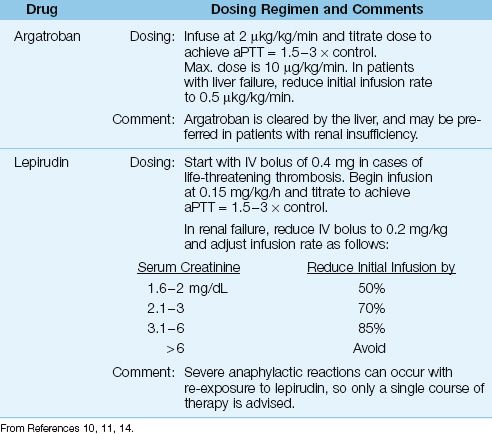
Acute Management
Heparin must be discontinued immediately (don’t forget to discontinue heparin flushes and remove heparin-coated catheters). Therapeutic anticoagulation with one of the direct thrombin inhibitors shown in Table 19.2 should be started immediately, even in cases where HIT is not accompanied by thrombosis (11). The recommendation for full anticoagulation without evidence of thrombosis is based on studies showing a 10-fold higher incidence of thrombosis following the appearance of HIT when anticoagulation is delayed (13).
ARGATROBAN: Argatroban is a synthetic analogue of L-arginine that reversibly binds to the active site on thrombin. It has a rapid onset of action, and is given by continuous infusion using the dosing regimen in Table 19.2. The therapeutic goal is an activated partial thromboplastin time (aPTT) of 1.5 to 3 times control values. The drug is cleared primarily by the liver, and a dose adjustment is necessary in hepatic insufficiency. Argatroban is recommended in patients with renal insufficiency (11) because a dose adjustment is not necessary.
LEPIRUDIN: Lepirudin is a recombinant form of hirudin, an anticoagulant found in leech saliva (!) that binds irreversibly to thrombin. Lepirudin is also given by continuous infusion, which can be preceded by a bolus injection in cases of life-threatening thrombosis. The therapeutic goal is the same as with argatroban (aPTT=1.5–3 × control). Lepirudin is cleared by the kidneys, and a dose adjustment is necessary when renal function is even mildly impaired (i.e., when the serum creatinine is above 1.5 mg/dL), as shown in Table 19.2 (14). Using argatroban in patients with renal impairment will avoid dosing adjustments. Finally, re-exposure to lepirudin can produce life-threatening anaphylactic reactions (11), so the treatment of HIT with lepirudin is usually a one-time affair.
DURATION OF TREATMENT: Full anticoagulation with argatroban or lepirudin is recommended until the platelet count rises above 150,000/∝L (11). Thereafter, coumadin can be used for long-term anticoagulation if HIT is associated with thrombosis, but there are 2 caveats: (a) coumadin should NOT be started until the platelet count increases beyond 150,000/∝L, and (b) the initial coumadin dose should not exceed 5 mg (11). These precautions are intended to reduce the risk of limb gangrene associated with coumadin therapy during the active phase of HIT (as mentioned earlier). The antithrombin agents should be continued until coumadin achieves full anticoagulation.
Thrombotic Microangiopathies
A thrombotic microangiopathy is a clinical disorder with the following features:
1. Widespread microvascular thrombosis with multiorgan dysfunction or failure.
2. A consumptive thrombocytopenia.
3. Fragmentation of erythrocytes in the clot-filled microvasculature, resulting in a microangiopathic hemolytic anemia.
These features are identified in the following clinical disorders:
A. Disseminated intravascular coagulation (DIC).
B. Thrombotic thrombocytopenia purpura (TTP).
C. The HELLP syndrome: hemolysis, elevated liver enzymes, and low platelets.
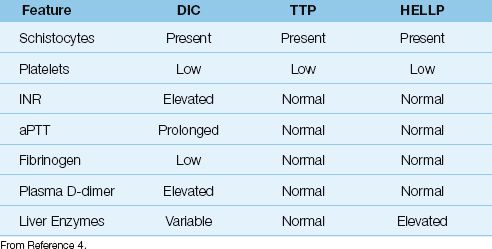
The comparative features of these 3 conditions are shown in Table 19.3.
Table 19.3 Comparative Features of the Thrombotic Microangiopathies
Disseminated Intravascular Coagulation
Disseminated intravascular coagulation (DIC) is a secondary disorder that is triggered by conditions that produce widespread tissue injury such as multisystem trauma, severe sepsis and septic shock, and obstetric emergencies (amniotic fluid embolism, abruptio placentae, eclampsia, and retained fetus syndrome). The inciting event is release of tissue factor, which (as described earlier) activates a series of clotting factors in the bloodstream that culminates in the formation of fibrin. This leads to widespread microvascular thrombosis and secondary depletion of platelets and clotting factors, resulting in a consumptive coagulopathy (15).
Clinical Features
The microvascular thrombosis in DIC can lead to multiorgan failure, most often involving the lungs, kidneys, and central nervous system, while depletion of platelets and coagulation factors can promote bleeding, particularly from pre-existing lesions in the GI tract such as stress ulcers. DIC can also be accompanied by symmetrical necrosis and ecchymosis involving the limbs, a condition known as purpura fulminans that is usually seen with overwhelming systemic sepsis, most notably with meningococcemia (7).
HEMATOLOGIC ABNORMALITIES: In addition to thrombocytopenia, DIC is usually (but not always) associated with elevation of the INR (i.e., prolongation of the prothrombin time) and prolongation of the activated partial thromboplastin time (aPTT), both abnormalities being the result of consumption and subsequent depletion of clotting factors in blood. The enhanced thrombosis is also accompanied by enhanced fibrinolysis, which elevates the fibrin degradation products in plasma (i.e., plasma D-dimers). Finally, the microangiopathic hemolytic anemia is identified by the presence of damaged or fragmented erythrocytes in a peripheral blood smear, like the ones in Figure 19.1. The fragmented erythrocytes are known as schistocytes, and they are the hallmark of the thrombotic microangiopathies.
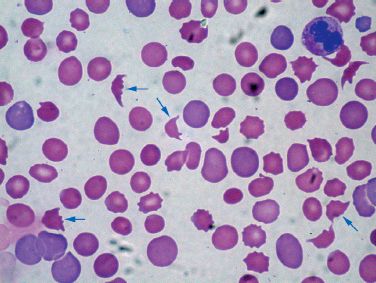
FIGURE 19.1 Peripheral blood smear from a patient with DIC. The arrows are pointing to schistocytes, which are fragmented erythrocytes. Their presence in a peripheral blood smear confirms the presence of a microangiopathic hemolytic anemia.
Management
There is no specific treatment for DIC other than supportive care. Uncontrolled bleeding often prompts consideration of replacement therapy with platelets and coagulation factors (plasma products), but this rarely helps and can be deleterious by “adding fuel” to the microvascular thrombosis. In severe cases of DIC associated with multiorgan failure, the mortality rate is 80% or higher (7,15).
Thrombotic Thrombocytopenia Purpura
Thrombotic thrombocytopenia purpura (TTP) is thrombotic microangiopathy that is caused by platelet binding to abnormal von Willebrand factor on microvascular endothelium (4). This can be a devastating condition that is fatal within 24 hours of onset. There is often no predisposing condition, although it seems to follow a nonspecific viral illness in some cases.
Clinical Features
TTP presents with a characteristic pentad of clinical manifestations that includes fever, altered mental status, acute renal failure, thrombocytopenia, and microangiopathic hemolytic anemia. The presence of all 5 conditions is not necessary for the diagnosis of TTP, but the diagnosis does require thrombocytopenia and evidence of a microangiopathic hemolytic anemia (e.g., schistocytes in the peripheral blood smear). TTP can be distinguished from DIC because clotting factors are not depleted in TTP, so the INR, aPTT, and fibrinogen levels are normal in TTP.
Management
Platelet transfusions are contraindicated in TTP because they can aggravate the underlying thrombosis. The treatment of choice for TTP is plasma exchange (16,17), where blood from the patient is diverted to a device that separates and discards the patient’s plasma and reinfuses plasma from a healthy donor. This is continued until 1.5 times the normal plasma volume is exchanged, and this process is repeated daily for 3–7 days. Acute fulminant TTP is almost always fatal if untreated, but if plasma exchange is started early (with 48 hours of symptom onset), as many as 90% of patients can survive the illness (16,17).
HELLP Syndrome
HELLP (Hemolysis, Elevated Liver enzymes, Low Platelets) syndrome is a thrombotic microangiopathy that occurs late in pregnancy or in the early postpartum period (18). About 20% of cases are associated with severe pre-eclampsia, and there is also an association with the antiphospholipid syndrome (19). The culprit in the HELLP syndrome is unexplained activation of clotting factors and platelets leading to microvascular thrombosis. There is also an unexplained elevation in liver enzymes, principally the transaminases (18).
Clinical Features
As the name indicates, HELLP is identified by the characteristic triad of hemolysis, thrombocytopenia, and elevated liver enzymes. HELLP can be confused with DIC (which can occur in the same clinical settings), but the INR and aPTT are usually normal in HELLP because there is no depletion of clotting factors, and this feature should distinguish HELLP from DIC (see Table 19.3).
The HELLP syndrome is an obstetric emergency, and a detailed description of this condition is beyond the scope of this text. For more information on HELLP, some recent reviews are included in the bibliography at the end of the chapter (18,19).
PLATELET TRANSFUSIONS
Platelet Products
Platelets are obtained either by pooling the platelets from multiple donors, or by extracting platelets from a single donor using apheresis techniques.
Pooled Platelets
Platelets are separated from fresh whole blood by differential centrifugation, and the resulting platelet concentrates from 5 units of whole blood (from 5 individual donors) are pooled together prior to storage. The pooled platelet concentrate contains about 38 × 1010 platelets in 260 mL plasma, which is equivalent to a platelet count of about 130 × 109/∝L. This is six orders of magnitude higher than the normal platelet count in blood (150–400 × 103/∝L). Platelets are stored at 20–24°C, and can be stored for up to 5 days.
Apheresis Platelets
Apheresis platelets are collected from a single donor and have a platelet count and volume that is equivalent to the pooled platelets from 5 donors. The presumed benefit of single-donor platelet transfusions is a lower risk of transmitted infections and a lower incidence of platelet alloimmunization (i.e., developing antibodies to donor platelets). How-ever, neither of these proposed benefits has been documented in clinical trials (20), and when leukocytes are removed from platelet products, there is no difference in the risk of platelet alloimmunization with single-donor and multiple-donor platelet transfusions (22).
Leukoreduction
Leukocytes in donor blood have been implicated in several adverse reactions, and leukocyte removal using specialized filters is now a routine practice for erythrocyte transfusions (see last chapter). Platelet concentrates are not free of leukocytes, and leukocyte reduction for platelet transfusions has the following advantages (20,22): a lower incidence of cytomegalovirus transmission (because this organism is transmitted in leukocytes), fewer febrile reactions, and a lower incidence of platelet alloimmunization. Because of these advantages, leukocyte reduction is becoming a routine practice for platelet transfusions.
Response to Transfused Platelets
In an average sized adult with no ongoing blood loss, a platelet concentrate from one unit of whole blood should raise the circulating platelet count by 7,000 to 10,000/∝L at one hour post-transfusion (20). Since an average of 5 platelet concentrates are pooled together for each platelet transfusion, the expected (or ideal) increase in platelet count is 35,000 to 50,000/∝L at one hour post-transfusion. The increment is about 40% lower after 24 hours, as shown in Figure 19.2. Note: The number of platelet concentrates that are pooled together can differ slightly (by ±1 unit) in individual platelet transfusions, so if you want to be precise about estimating post-transfusion platelet increments (which is usually not necessary), you need to enquire about the number of units of platelets included in the transfusion pack.
Multiple Transfusions
The increment in platelet count declines with multiple transfusions. This is shown in Figure 19.2, where the platelet increment is about 25% lower after 5 platelet transfusions (23). As mentioned earlier, this phenomenon of “platelet refractoriness” is the result of antiplatelet antibodies in the recipient directed at ABO antigens on donor platelets. This effect can be mitigated by transfusing ABO-matched platelets.
Indications for Platelet Transfusions
Active Bleeding
In the presence of active bleeding other than ecchymoses or petechiae, platelet transfusions are recommended to maintain a platelet count >50,000/∝L (21). For intracranial hemorrhage, higher platelet counts (>100,000/∝L) should be maintained (21).
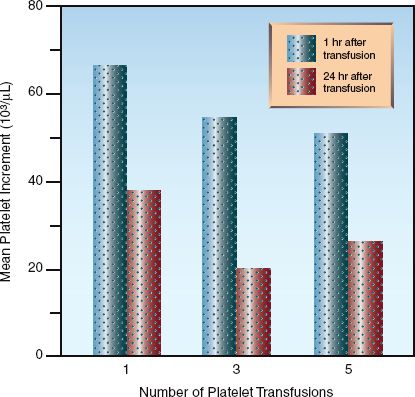
FIGURE 19.2 Post-transfusion increment in platelet counts in relation to the time elapsed after the transfusion (one hour versus 24 hours) and the number of transfusions given. Data from Reference 23.
No Active Bleeding
Despite evidence that spontaneous bleeding through an intact vascular system is uncommon with platelet counts down to 5,000/∝L (20), most experts are reluctant to adopt a platelet transfusion trigger as low as 5,000/∝L. In the absence of bleeding (other than ecchymoses or petechiae), prophylactic platelet transfusions are usually recommended when the platelet count reaches 10,000/∝L (21).
Procedures
In the absence of associated coagulation abnormalities:
1. Platelet counts >40,000/∝L are sufficient to perform laparotomy, craniotomy, tracheotomy, percutaneous liver biopsy, and bronchoscopic or endoscopic biopsy (20).
2. Platelet counts >20,000/∝L are sufficient to perform lumbar punctures (20).
3. Platelet counts >10,000/∝L are sufficient to perform central venous cannulation safely (24,25).
Adverse Effects
Bacterial Transmission
Bacteria are much more likely to flourish in platelet concentrates than in RBC concentrates (packed cells) because platelets are stored at room temperature (22°C), while RBCs are refrigerated at about 4°C. It is estimated that one in every 2,000 to 3,000 platelet concentrates harbors bacteria, and that one in 5,000 concentrates will produce sepsis in the recipient (16). Cultures of all platelet concentrates are now required (16) but, since platelets can be stored for only 5 days, the platelets can be transfused before the culture results are available.
Fever
Febrile nonhemolytic reactions have been reported in as many as 30% of platelet transfusions (26), which is much greater than the 0.5% rate of similar reactions reported with RBC transfusions (see Table 18.3 on page 359). Part of this difference may be related to the multiple donors used for platelet transfusions. Since antibodies to donor leukocytes are implicated in this reaction, leukoreduction of platelet products will help to alleviate this problem.
Hypersensitivity Reactions
Hypersensitivity reactions (urticaria, anaphylaxis, anaphylactic shock) are also more common with platelet transfusions that with erythrocyte transfusions (20). Since this is a reaction to proteins in donor plasma, removing the plasma from platelet concentrates will reduce the risk of hypersensitivity transfusion reactions.
Acute Lung Injury
Transfusion-related acute lung injury (TRALI) is described in Chapter 18 (see pages 361–363). This condition, which is an inflammatory lung injury similar to the acute respiratory distress syndrome (ARDS), is most often associated with erythrocyte transfusions, but has also been reported in association with platelet transfusions (27). The culprit is believed to be antileukocyte antibodies in donor blood that activate neutrophils in the recipient. TRALI usually appears within 6 hours after the start of a transfusion, and management is supportive.
PLASMA PRODUCTS
Plasma products are used as a source of coagulation factors, but surveys indicate that about 50% of plasma transfusions are inappropriate (28).
Fresh Frozen Plasma
Plasma is separated from donor blood and frozen at –18°C within 8 hours of blood collection. This fresh frozen plasma (FFP) has a volume of about 230 mL, and can be stored for one year. Once thawed, FFP can be stored at 1 to 6°C for up to 5 days. The principal uses of FFP include the resuscitation of massive blood loss, and the reversal of excess anticoagulation with coumadin.
Massive Blood Loss
As described in Chapter 11 (see pages 209–210), the use of FFP in massive blood loss (blood loss equivalent to one blood volume within 24 hours) has become more aggressive in recent years, mostly as a result of experiences in combat injuries. Whereas the traditional practice was to transfuse one unit of FFP for every 6 units of packed RBCs to prevent a dilutional coagulopathy, there is now evidence that severe trauma is accompanied by a coagulopathy (30), and survival rates have improved with FFP:RBC ratios of 1:2 to 1:3 during massive transfusion (31,32). This approach is called hemostatic resuscitation, and the goal is an INR that remains below 1.5. (Note: the international normalized ratio or INR is the ratio of the patient’s prothrombin time and an international standard for the normal or control prothrombin time; i.e., INR = Patient’s PT/standardized control PT.)
Warfarin-Induced Hemorrhage
The annual incidence of major bleeding during warfarin anticoagulation is 3 to 12%, and the incidence of fatal hemorrhage is 1 to 3% (33), with intracerebral hemorrhage causing most of the fatalities. The management of major or life-threatening hemorrhage related to excessive warfarin anticoagulation is shown in Table 19.4 (34,35). Since warfarin acts by inhibiting vitamin K-dependent clotting factors (i.e., factors II, VII, IX, X), vitamin K is administered to block ongoing anticoagulant activity. Clotting factors are then replenished, and this has traditionally involved the infusion of FFP at a volume of 15 mL/kg. There are two shortcomings with the use of FFP in this setting: the time to normalize the INR can be prolonged, and the volume of fluid required can aggravate the bleeding. These problems can be mitigated by using the plasma product described next.
PROTHROMBIN COMPLEX CONCENTRATE: Normalizing the INR quickly with a limited infusion volume is possible with prothrombin complex concentrates (PCC) using the dosing recommendations in Table 19.4. There are 3-factor and 4-factor PCC preparations (the name indicating the number of vitamin K-dependent clotting factors in the preparation), but only the 3-factor PCC is approved for clinical use. PCC is a lyophilized powder that is quickly reconstituted, thereby avoiding the time delays involved in thawing FFP. Whereas FFP can take hours to normalize the INR, PCC can accomplish this task in less than 30 minutes (34,35). The rapid response and limited volume associated with PCC make it well suited for the treatment of warfarin-induced bleeding, particularly intra-cerebral hemorrhage (35).
Table 19.4 Management of Warfarin-Induced Hemorrhage

Cryoprecipitate
When FFP is allowed to thaw at 4°C, a milky residue forms that is rich in cold-insoluble proteins (cryoglobulins) like fibrinogen, von Willebrand factor, and factor VIII. This cryoprecipitate can be separated from plasma and stored at –18°C for up to one year. The storage volume is 10 to 15 mL.
Cryoprecipitate was introduced in 1965 as a concentrated source of factor VIII for the management of hemophilia, but it has been replaced by recombinant factor VIII preparations. The current use of cryoprecipitate in the ICU is limited to uncontrolled uremic bleeding and selected cases of hypofibrinogenemia.
Uremic Bleeding
Platelet adhesion is impaired in renal failure (acute and chronic) as a result of abnormal platelet binding to fibrinogen and von Willebrand factor (which anchors the platelet plug to the endothelium, as mentioned previously). Bleeding times are prolonged when the serum creatinine climbs above 6 mg/dL, and dialysis corrects the bleeding time in only 30 to 50% of patients (36).
The significance of the impaired platelet adhesiveness in renal failure is unclear. However, upper GI bleeding is the second leading cause of death in acute renal failure (36), so there is reason to be concerned about this platelet function abnormality. There are two treatment options for uremic bleeding: desmopressin and cryoprecipitate.
DESMOPRESSIN: Desmopressin is a vasopressin analogue (deamino-arginine vasopressin or DDAVP) that does not have the vasoconstrictor or antidiuretic effects of vasopressin, but is capable of elevating plasma levels of von Willebrand factor and correcting the abnormal bleeding time in 75% of patients with renal failure (36,37). The recommended dose is 0.3 ∝g/kg IV or by subcutaneous injection, or 30 ∝g/kg by intranasal spray (36,37). The effect lasts only 6 to 8 hours, and repeat dosing leads to tachyphylaxis.
Although desmopressin can correct the bleeding time in renal failure, the effect on uremic bleeding is not known. When uremic bleeding is worrisome, desmopressin can be given empirically in one or two doses (6 to 8 hours apart). If the bleeding persists, cryoprecipitate can be given (because it is rich in fibrinogen and von Willebrand factor, which are both involved in the platelet function abnormality in renal failure). The standard dose of cryoprecipitate for uremic bleeding is 10 units.
Hypofibrinogenemia
Cryoprecipitate can also be used as a source of fibrinogen in bleeding episodes associated with fibrinogen deficiency, such as variceal bleeding from liver failure. One unit of cryoprecipitate contains about 200 mg of fibrinogen, and infusion of 10 units of cryoprecipitate (2 grams of fibrinogen) should raise the serum fibrinogen level by about 70mg/dL in an average sized adult (38). The goal is a serum fibrinogen level above 100 mg/dL.
Adverse Effects
The risks associated with transfusion of plasma products are essentially the same risks associated with transfusion of erythrocytes and/or platelets. The exception is nonhemolytic febrile transfusion reactions, which are caused by donor leukocytes and thus should not occur with plasma transfusions.
Acute Hemolytic Reactions
Acute hemolytic reactions are caused by anti-A and anti-B antibodies in the transfused plasma that react with A and B antigens on recipient RBCs. Since cross-matching of plasma transfusions is not a universal practice, acute hemolytic reactions continue to be reported with plasma transfusions. The evaluation of suspected acute hemolytic transfusion reactions is described in Chapter 18 (see pages 359–360).
Transmitted Infections
Plasma transfusions carry a minimal risk of transmitting infections. The risk of hepatitis B transmission is 1 per 900,000 transfusions, the risk of hepatitis C transmission is 1 per 30 million transfusions, and the risk of HIV transmission is one per 8 million transfusions (39). The risk of bacterial transmission is reported as “rare” and CMV transmission, which occurs in transfused leukocytes, has not been reported with plasma transfusions (39).
Hypersensitivity Reactions
Hypersensitivity reactions (urticaria, anaphylaxis, anaphylactic shock), which are caused by sensitization to proteins in donor plasma, are more common with plasma transfusions than with erythrocyte or platelet transfusions. These reactions are, however, uncommon; e.g., the reported incidence of allergic reactions in the United Kingdom is approximately 1 case per 17,000 plasma transfusions (39).
Acute Lung Injury
Transfusion-related acute lung injury (TRALI) is attributed to antileukocyte antibodies in donor blood, and is a complication of erythrocyte, platelet, and plasma transfusions. The reported incidence of TRALI after platelet transfusions is 1 per 60,000 units (39), which is much less than the reported incidence following RBC transfusions (1 per 12,000 units). The clinical features of TRALI are described in Chapter 18 (see pages 361–363).
A FINAL WORD
The following points in this chapter deserve emphasis.
1. The transfusion of platelets or plasma is rarely indicated in the absence of active bleeding. In fact, in most life-threatening cases of thrombocytopenia (e.g., HIT, DIC, TTP, HELLP), the major problem is thrombosis, not hemorrhage.
2. The presence of a coagulopathy is not an absolute contraindication to inserting central venous catheters, even with platelet counts as low as 10,000/∝L.
3. If heparin-induced thrombocytopenia is suspected, don’t forget to remove heparin from catheter flushes, and to remove heparin-coated catheters.
4. For the management of major hemorrhage associated with warfarin anticoagulation, prothrombin complex concentrate is superior to fresh frozen plasma for correcting the coagulopathy, especially if the bleeding is intracranial.
REFERENCES
Hemostasis
1. King KE (ed). Overview of hemostasis. In: Blood transfusion therapy: A physician’s handbook. 9th ed. Bethesda, MD: American Association of Blood Banks, 2008.
2. Wheeler AP, Rice TW. Coagulopathy in critically ill patients. Part 2 – Soluble clotting factors and hemostatic testing. Chest 2010; 137:185–194.
Thrombocytopenia in the ICU
3. Parker RI. Etiology and significance of thrombocytopenia in critically ill patients. Crit Care Clin 2012; 28:399–411.
4. Rice TR, Wheeler RP. Coagulopathy in critically ill patients. Part 1:Platelet disorders. Chest 2009; 136:1622–1630.
5. Slichter SJ, Harker LA. Thrombocytopenia: mechanisms and management of defects in platelet production. Clin Haematol 1978; 7:523–527.
6. Payne BA, Pierre RV. Pseudothrombocytopenia: a laboratory artifact with potentially serious consequences. Mayo Clin Proc 1984; 59:123–125.
7. DeLoughery TG. Critical care clotting catastrophes. Crit Care Clin 2005; 21:531–562.
8. Francois B, Trimoreau F, Vignon P, et al. Thrombocytopenia in the sepsis syndrome: role of hemophagocytosis and macrophage colony-stimulating hormone. Am J Med 1997; 103:114–120.
9. Priziola JL, Smythe MA, Dager WE. Drug-induced thrombocytopenia in critically ill patients. Crit Care Med 2010; 38(Suppl):S145–S154.
10. Shantsila E, Lip GYH, Chong BH. Heparin-induced thrombocytopenia: a contemporary clinical approach to diagnosis and management. Chest 2009; 135:1651–1664.
11. Linkins L-A, Dans AL, Moores LK, et al. Treatment and prevention of heparin-induced thrombocytopenia. Antithrombotic Therapy and Preven-tion of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(Suppl):495S–530S.
12. Laster J, Silver D. Heparin-coated catheters and heparin-induced thrombocytopenia. J Vasc Surg 1988; 7:667–672.
13. Greinacher A, Eichler P, Lubenow N, et al. Heparin-induced thrombocytopenia with thromboembolic complications: a meta-analysis of 2 prospective trials to assess the value of parenteral treatment with lepirudin and its therapeutic aPTT range. Blood 2000; 96:846–851.
14. Lepirudin drug monograph. In McEvoy GK, ed. AHFS Drug Information, 2012. Bethesda, MD: American Society of Health System Pharmacists, 2012:1476–1478.
15. Senno SL, Pechet L, Bick RL. Disseminated intravascular coagulation (DIC). Pathophysiology, laboratory diagnosis, and management. J Intensive Care Med 2000; 15:144–158.
16. Rock GA, Shumack KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenia purpura. N Engl J Med 1991; 325:393–397.
17. Hayward CP, Sutton DMC, Carter WH Jr, et al. Treatment outcomes in patients with adult thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Arch Intern Med 1994; 154:982–987.
18. Kirkpatrick CA. The HELLP syndrome. Acta Clin Belg 2010; 65:91–97.
19. Di Prima FAF, Valenti O, Hyseni E, et al. Antiphospholipid syndrome during pregnancy: the state of the art. J Prenat Med 2011; 5:41–53.
Platelet Transfusions
20. Slichter SJ. Platelet transfusion therapy. Hematol Oncol Clin N Am 2007; 21:697–729.
21. Slichter SJ. Evidence-based platelet transfusion guidelines. Hematol 2007; 2007:172–178.
22. The Trial to Reduce Alloimmunization to Patients Study Group. Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. N Engl J Med 1997; 337:1861–1869.
23. Slichter SJ, Davis K, Enright H, et al. Factors affecting post-transfusion platelet increments, platelet refractoriness, and platelet transfusion intervals in thrombocytopenic patients. Blood 2005; 105:4106–4114.
24. Doerfler ME, Kaufman B, Goldenberg AS. Central venous catheter placement in patients with disorders of hemostasis. Chest 1996; 110:185–188.
25. DeLoughery TG, Liebler JM, Simonds V, et al. Invasive line placement in critically ill patients: Do hemostatic defects matter? Transfusion 1996; 36:827–831.
26. Gelinas J-P, Stoddart LV, Snyder EL. Thrombocytopenia and critical care medicine. J Intensive Care Med 2001; 16:1–21.
27. Sayah DM. Looney MR, Toy P. Transfusion reactions. Newer concepts on the pathophysiology, incidence, treatment, and prevention of transfusion-related acute lung injury. Crit Care Clin 2012; 28:363–372.
Plasma Products
28. Lauzier F, Cook D, Griffith L, et al. Fresh frozen plasma transfusion in critically ill patients. Crit Care Med 2007; 35:1655–1659.
29. Roback JD, Caldwell S, Carson J, et al. Evidence-based practice guidelines for plasma transfusion. Transfusion 2010; 50:1227–1239.
30. Brohi K, Singh J, Heron M, Coats T. Acute traumatic coagulopathy. J Trauma 2003; 54:1127–1130.
31. Beekley AC. Damage control resuscitation: a sensible approach to the exsanguinating surgical patient. Crit Care Med 2008; 36:S267–S274.
32. Magnotti LJ, Zarzaur BL, Fischer PE, et al. Improved survival after hemostatic resuscitation: does the emperor have no clothes? J Trauma 2011; 70:97–102.
33. Landefeld CS, Goldman L. Major bleeding in outpatients treated with warfarin: incidence and prediction by factors known at the start of outpatient therapy. Ann Intern Med 1989; 87:144–152.
34. Zareh M, Davis A, Henderson S. Reversal of warfarin-induced hemorrhage in the emergency department. West J Emerg Med 2011; 12:386–392.
35. Imberti D, Barillari G, Biasioli C, et al. Emergency reversal of anticoagulation with a three-factor prothrombin complex concentrate in patients with intracerebral hemorrhage. Blood Transfus 2011; 9:148–155.
36. Salman S. Uremic bleeding: pathophysiology, diagnosis, and management. Hosp Physician 2001; 37:45–76.
37. Mannucci PM. Desmopressin (DDAVP) in the treatment of bleeding disorders. The first 20 years. Blood 1997; 90:2515–2521.
38. Callum JL, Karkouti K, Lin Y. Cryoprecipitate: the current state of knowledge. Transfus Med Rev 2009; 23:177–184.
39. MacLennan S, Williamson LM. Risks of fresh frozen plasma and platelets. J Trauma 2006; 60(Suppl):546–550.







Full access? Get Clinical Tree


