FIGURE 15.1 Flow diagram for the evaluation of tachycardias.
Irregular Rhythm
If the R-R intervals are not uniform in length (indicating an irregular rhythm), the most likely arrhythmias are multifocal atrial tachycardia and atrial fibrillation. Once again, the atrial activity on the ECG helps to identify each of these rhythms; i.e.,
1. Multiple P wave morphologies with variable P-R intervals is evidence of multifocal atrial tachycardia (see Panel A, Figure 15.3).
2. The absence of P waves with highly disorganized atrial activity (fibrillation waves) is evidence of atrial fibrillation (see Panel B, Figure 15.3).
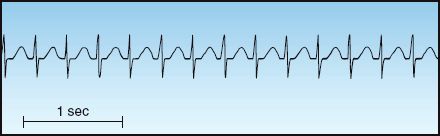
FIGURE 15.2 Narrow-QRS-complex tachycardia with a regular rhythm. Note the absence of P waves, which are hidden in the QRS complexes. This is an AV nodal re-entrant tachycardia.
Wide-QRS-Complex Tachycardias
Tachycardias with a wide QRS complex (>0.12 sec) can originate from a site below the AV conduction system (i.e., ventricular tachycardia), or they can represent a supraventricular tachycardia (SVT) with prolonged AV conduction (e.g., from a bundle branch block). These two arrhythmias can be difficult to distinguish. An irregular rhythm is evidence of an SVT with aberrant AV conduction, while certain ECG abnormalities (e.g., AV dissociation) provide evidence of VT. The distinction between VT and SVT with aberrant conduction is described in more detail later in the chapter.
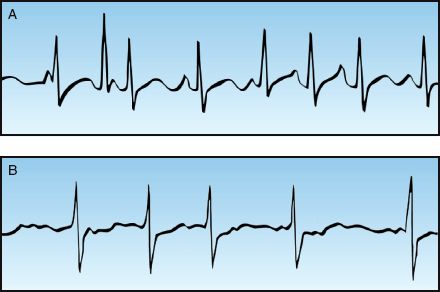
FIGURE 15.3 Narrow-QRS-complex tachycardias with an irregular rhythm. Panel A shows a multifocal atrial tachycardia (MAT), identified by multiple P wave morphologies and variable PR intervals. Panel B is atrial fibrillation, identified by the absence of P waves and the highly disorganized atrial activity (fibrillation waves).
ATRIAL FIBRILLATION
Atrial fibrillation (AF) is the most common cardiac rhythm disturbance in clinical practice, and can be paroxysmal (resolves spontaneously), recurrent (2 or more episodes), persistent (present for at least 7 days), or permanent (present for at least one year) (1,2). Most patients with AF are elderly (median age 75 years) and have underlying cardiac disease. About 25% of patients are less than 60 years of age and have no underlying cardiac disease (1): a condition known as lone atrial fibrillation.
Postoperative AF
Postoperative AF is reported in up to 45% of cardiac surgeries, up to 30% of non-cardiac thoracic surgeries, and up to 8% of other major surgeries (5). It usually appears in the first 5 postoperative days (6), and is associated with longer hospital stays and increased mortality (5,6). Several predisposing factors have been implicated, including heightened adrenergic activity, magnesium depletion, and oxidant stress. Prophylaxis with β-blockers and magnesium is currently popular (5,7), and there is evidence that the antioxidant N-acetylcysteine (a glutathione surrogate) provides effective prophylaxis following cardiac surgery (7). Most cases of postoperative AF resolve within a few months.
Adverse Consequences
The adverse consequences of AF include impaired cardiac performance and thromboembolism.
Cardiac Performance
Atrial contraction is responsible for 25% of the ventricular end-diastolic volume in the normal heart (8). Loss of the atrial contribution to ventricular filling in AF has little noticeable effect when cardiac function is normal, but it can result in a significant decrease in stroke output when diastolic filling is impaired by mitral stenosis or reduced ventricular compliance (1). This effect is more pronounced at rapid heart rates (because of the reduced time for ventricular filling).
Thromboembolism
Atrial fibrillation predisposes to thrombus formation in the left atrium, and these thrombi can dislodge and embolize in the cerebral circulation, thereby producing an acute ischemic stroke. The average yearly incidence of ischemic stroke is 3 to 5 times higher in patients with AF, but only when the AF is accompanied by certain risk factors (e.g., heart failure, mitral stenosis, advanced age) (1,2). Recommendations for antithrombotic therapy are presented later.
Management Strategies
The acute management of AF can be divided into 3 components: i.e., control of the heart rate, cardioversion (electrical and pharmacological), and thromboprophylaxis.
Heart Rate Control
The typical strategy for uncomplicated AF is to slow the ventricular response with drugs that prolong AV conduction. A variety of drugs are available for this purpose, and the popular ones are included in Table 15.1. The following is a brief description of these drugs.
Table 15.1 Drug Regimens for Acute Rate Control in Atrial Fibrillation
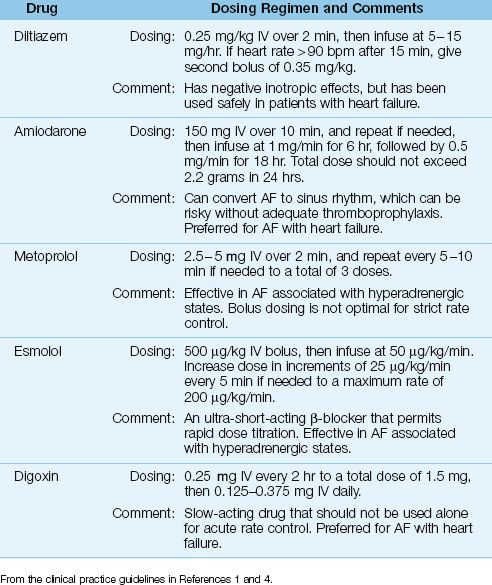
DILTIAZEM: Diltiazem is a calcium channel blocker that achieves satisfactory rate reduction in up to 90% of cases of uncomplicated AF (9). The acute response to diltiazem is shown in Figure 15.4: note the superiority of diltiazem over amiodarone and digoxin after the first hour of therapy. Adverse effects of diltiazem include hypotension and cardiac depression. Although diltiazem has negative inotropic effects, it has been used safely in patients with moderate to severe heart failure (10).
β-RECEPTOR ANTAGONISTS: β-blockers achieve successful rate control in 70% of cases of acute AF (11), and they are the preferred agents for rate control when AF is associated with hyperadrenergic states (such as acute MI and post-cardiac surgery) (1,5). Two β-blockers with proven efficacy in AF are esmolol (Brevibloc) and metoprolol (Lopressor). Both are cardioselective agents that preferentially block β-1 receptors in the heart. Esmolol is more attractive than metoprolol because it is an ultra short-acting drug (with a serum half-life of 9 minutes), which allows rapid dose titration to the desired effect (12).
AMIODARONE: Amiodarone prolongs conduction in the AV node, but is less effective for acute rate control than diltiazem, as shown in Figure 15.4. However, amiodarone produces less cardiac depression than diltiazem (13), and it is favored by some for AF with heart failure (1). Amiodarone is also an antiarrhythmic agent (Class III), and is capable of converting AF to a sinus rhythm. The success rate for converting recent-onset AF is 55% to 95% when a loading dose and continuous infusion are used, and the daily dose exceeds 1500 mg (1,14). However, unanticipated cardioversion with amiodarone can be a problem when patients are not adequately anticoagulated (see later).
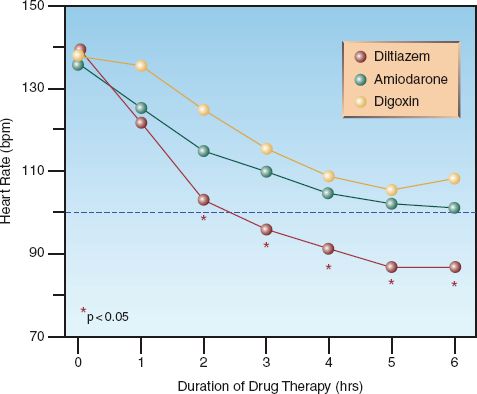
FIGURE 15.4 Comparison of acute rate control with intravenous diltiazem, amiodarone, and digoxin in patients with uncomplicated atrial fibrillation. Data points marked with a star indicate a significant difference with diltiazem compared to the other two drugs. Data from Reference 10.
The adverse effects of short-term intravenous amiodarone include hypo-tension (15%), infusion phlebitis (15%), bradycardia (5%), and elevated liver enzymes (3%) (15,16). Hypotension is the most common side effect, and is related to the combined vasodilator actions of amiodarone and the solvent (polysorbate 80 surfactant) used to enhance water solubility of injectable amiodarone (17). Amiodarone also has several drug interactions by virtue of its metabolism by the cytochrome P450 enzyme system in the liver (16). The most relevant interactions in the ICU setting are inhibition of digoxin and warfarin metabolism, which requires attention if amiodarone is continued orally for long-term management.
DIGOXIN: Digoxin prolongs conduction in the AV node, and is a popular drug for long-term rate control in AF. However, the response to intravenous digoxin is slow to develop; i.e., it is usually not apparent for at least one hour, and the peak response can take longer than 6 hours to develop (1). In comparison, the response to an intravenous dose of diltiazem is apparent in 3–5 minutes, and the peak response occurs at 5–7 minutes (17). The superiority of diltiazem over digoxin for acute rate control in AF is demonstrated in Figure 15.4. Note that the heart rate remains above 100 beats/min (the threshold for tachycardia) at 6 hours after the start of rate-control therapy with digoxin. Digoxin may have a role in treating AF associated with heart failure, but it should not be used alone for acute rate control in AF (1,4).
Electrical Cardioversion
More than 50% of episodes of recent-onset AF will spontaneously revert to sinus rhythm within the first 72 hours (18). For the remaining cases of AF that are complicated by hypotension, pulmonary edema, or myocardial ischemia, direct-current cardioversion is the appropriate intervention. Biphasic shocks have replaced monophasic shocks as the standard of care for cardioversion because they require less energy for a successful result. An energy of 100 J is usually enough for successful cardioversion using biphasic shocks, but 200 J is recommended for the initial cardioversion attempt in the most recent guidelines on AF (1). If additional shocks are needed, the energy level is increased in increments of 100 J to a maximum energy level of 400 J. Success may be fleeting when AF has persisted for more than a year (1).
Pharmacological Cardioversion
Drug-induced cardioversion is used in cases of uncomplicated AF that are refractory to rate control, or for first episodes of uncomplicated AF that are less than 48 hours in duration, to eliminate the need for anticoagulation (see later). Several antiarrhythmic agents can be effective in terminating AF, including amiodarone and ibutilide. The success rate of amiodarone in recent-onset AF has been mentioned previously. Ibutilide (at a dose of 1 mg IV over 10 minutes, repeated once if necessary) has a reported success rate of about 50% in recent-onset AF (15). Ibutilide prolongs the QT interval, and is one of the high-risk drugs for promoting polymorphic ventricular tachycardia (torsade de pointes) (15), as shown later in Table 15.4
Thromboprophylaxis
Recommendations for antithrombotic therapy in AF are shown in Table 15.2. To summarize, anticoagulation is recommended for any patient with AF and one or more of the following risk factors: mitral stenosis, coronary artery disease, congestive heart failure, hypertension, age ≥75 yrs, diabetes mellitus, and prior stroke or TIA (1,2). This, of course, excludes patients who have a contraindication to anticoagulation.
Table 15.2 Antithrombotic Therapy in Atrial Fibrillation (AF)
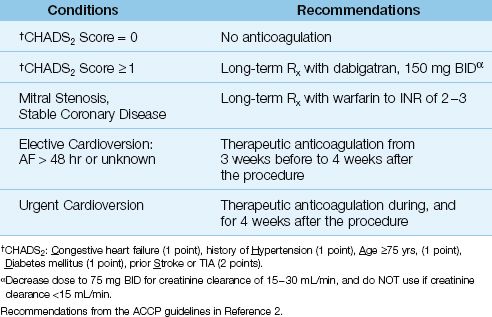
DABIGATRAN: The ACCP guidelines (2) recommend the use of dabigatran (150 mg BID), a direct thrombin inhibitor, for patients with AF and one or more risk factors in the CHADS2 score. This is based on one study showing that dabigatran in a dose of 150 mg BID (but not lower) was associated with fewer strokes than warfarin (19). This study excluded patients with renal insufficiency because dabigatran is cleared by the kidneys. In fact, it is important to note that a dose reduction of 50% (to 75 mg BID) is required for patients with renal impairment (i.e., creatinine clearance of15–30 mL/min), while the drug is contraindicated in patients with renal failure (i.e., creatinine clearance <15 mL/min) (2).
Since the degree of anticoagulation is not routinely monitored when using thrombin inhibitors, and there is no antidote to reverse dabigatran in cases of drug-associated bleeding, it is probably wise to avoid this drug in patients with any degree of renal impairment.
CARDIOVERSION: Thromboembolic events following cardioversion have been reported in 1% to 7% of patients who were not anticoagulated at the time of the procedure (1,2). This is the basis for the recommendation to begin anticoagulation 3 weeks prior to an elective cardioversion, and continue anticoagulation for 4 weeks after the procedure (2). For emergent cardioversion, anticoagulation with heparin should be initiated as soon as possible prior to the procedure, followed by four weeks of anticoagulation after the procedure. When AF is less than 48 hours in duration, cardioversion has a low risk of thromboembolism (<1%), and peri-procedural anticoagulation is not necessary (1).
Wolff-Parkinson-White Syndrome
The Wolff-Parkinson-White (WPW) syndrome (short P–R interval and delta waves before the QRS) is characterized by recurrent supraventricular tachycardias that originate from an accessory pathway in the AV node. (The mechanism for these tachycardias is explained in the section on re-entrant tachycardias.) When atrial fibrillation occurs in a patient with an accessory pathway, drugs that block conduction in the AV node (e.g., calcium channel blockers, β-blockers, digoxin) are unlikely to slow the ventricular rate because the accessory pathway is not blocked (1,4). Furthermore, selective block of the AV node can precipitate ventricular fibrillation (4). Therefore, drugs that block the AV node (e.g., calcium channel blockers, β-blockers, and digoxin) should NOT be used when AF is associated with the WPW syndrome (1,4). The preferred management in this situation is electrical cardioversion or antiarrhythmic drugs such as amiodarone or procainamide.
MULTIFOCAL ATRIAL TACHYCARDIA
Multifocal atrial tachycardia or MAT (see Panel A in Figure 15.3) is a disorder of the elderly (average age = 70), and over half of the cases occur in patients with chronic lung disease (20). Other associated conditions include magnesium and potassium depletion, and coronary artery disease (21).
Acute Management
The following measures are recommended for the acute management of MAT, but this is a stubborn arrhythmia that is often refractory to medical management.
1. Identify and correct hypomagnesemia and hypokalemia if necessary. If both disorders co-exist, the magnesium deficiency must be corrected before potassium deficits can be replaced. This is ex-plained in Chapter 37.
2. Since serum magnesium levels can be normal when total body magnesium is depleted (also explained in Chapter 37), intravenous magnesium can be given as an empiric measure when serum magnesium levels are normal. The following regimen can be used:
Start with 2 grams MgSO4 (in 50 mL saline) IV over 15 minutes,
then infuse 6 grams MgSO4 (in 500 mL saline) over 6 hours.
In one study, this regimen had a remarkable 88% success rate in converting MAT to a sinus rhythm, and the effect was independent of serum magnesium levels (21). This success may be explained by the membrane-stabilizing effect of magnesium (22), and by the actions of magnesium as “nature’s calcium channel blocker” (see Chapter 37). There is no risk of magnesium overload from this empiric regimen.
3. If the prior measures fail, and COPD is not the cause of the MAT, metoprolol in the doses shown in Table 15.1 has a reported 80% success rate in converting MAT to sinus rhythm (20). If metoprolol is a concern in patients with COPD, the calcium channel blocker verapamil can be effective. Verapamil converts MAT to sinus rhythm in less than 50% of cases (20), but it can also slow the ventricular rate. The dose is 0.25–5 mg IV over 2 min, which can be repeated every 15–30 min, if necessary, to a total dose of 20 mg (4). Verapamil is a potent negative inotropic agent, and hypotension is a frequent side effect. The drug is not recommended for patients with heart failure (4).
PAROXYSMAL SUPRAVENTRICULAR TACHYCARDIA
Paroxysmal supraventricular tachycardias (PSVT) are narrow-QRS-complex tachycardias that are second only to atrial fibrillation as the most prevalent rhythm disturbances in the general populace.
Mechanism
These arrhythmias occur when impulse transmission in one pathway of the AV conduction system is slowed. This creates a difference in the refractory period for impulse transmission in the abnormal and normal conduction pathways, and this allows impulses travelling down one pathway to travel back through the other pathway. The retrograde transmission of impulses is called re-entry, and it results in a circular pattern of impulse transmission that is self-sustaining; i.e., a re-entrant tachycardia. Re-entry is triggered by an ectopic atrial impulse in one of the two conduction pathways, which results in the abrupt onset that is characteristic of re-entrant tachycardias.
There are 5 different types of PSVT, based on the location of the re-entrant pathway. The most common PSVT is AV nodal re-entrant tachycardia, where the re-entrant pathway is located in the AV node.
AV Nodal Re-Entrant Tachycardia
AV nodal re-entrant tachycardia (AVNRT) accounts for 50 to 60% of cases of PSVT (23). It typically appears in patients who have no history of heart disease, and is more common in women than men. The onset is abrupt, and the predominant complaints are palpitations and lightheadedness. There is no evidence of heart failure or myocardial ischemia, and significant hemodynamic compromise is uncommon. The ECG shows a narrow-QRS-complex tachycardia with a regular rhythm and a heart rate between 140 and 250 bpm (23). There are often no visible P waves on the ECG, as shown in Figure 15.2.
AVNRT is frequently mistaken for sinus tachycardia, but the onset is different (abrupt onset vs. gradual onset for sinus tachycardia), the heart rate is often different (usually higher than 140 beats/min in AVNRT and rarely higher than 150 beats/min in sinus tachycardia), and the ECG appearance is different (no visible P waves in AVNRT and P waves before each QRS complex in sinus tachycardia).
Vagal Maneuvers
Maneuvers that increase vagal tone are recommended as an initial attempt to terminate AVNRT. A variety of maneuvers have been identified, including carotid sinus massage and the Valsalva maneuver (maximal expiratory effort with a closed glottis). The success of these maneuvers has not been adequately studied; one study of 148 patients with paroxysmal SVT showed a success rate of 18% with the Valsalva maneuver and 12% with carotid sinus massage (24).
Adenosine
When vagal maneuvers are ineffective, adenosine is the drug of choice for terminating re-entrant tachycardias involving the AV node (25,26). Adenosine is an endogenous nucleotide (the backbone of the ATP molecule) that relaxes vascular smooth muscle and slows conduction in the AV node. When given by rapid intravenous injection, adenosine has a rapid onset of action (<30 sec) and produces a transient AV block that can terminate AV nodal re-entrant tachycardias. Adenosine is quickly cleared from the bloodstream (by receptors on RBCs and endothelial cells), and the effects last only 1–2 minutes.
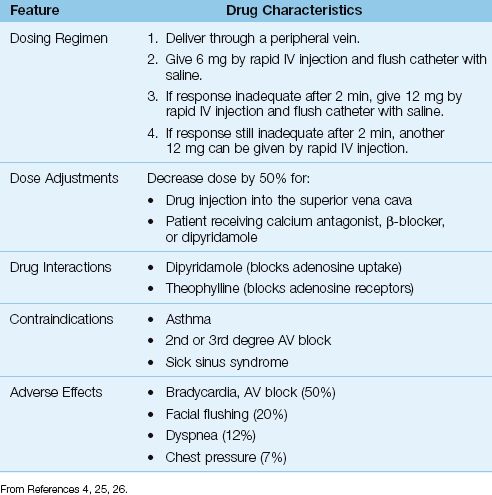
Dosing Considerations
The dosing regimen for adenosine is shown in Table 15.3. The initial dose is 6 mg, which is injected rapidly into a peripheral vein and followed by a saline flush. Optimal results are obtained if the drug is injected at the hub of the catheter. If conversion to sinus rhythm does not occur after 2 minutes, a second 12 mg dose is given, and this can be repeated once if necessary. This regimen terminates re-entrant tachycardias in over 90% of cases (24–26). The effective dose of adenosine was determined using drug injection into peripheral veins, and ventricular asystole has been reported when standard doses of adenosine are injected through central venous catheters (27). As a result, a dose reduction of 50% has been recommended by some (including the manufacturer) when adenosine is injected through a central venous catheter (27).
Table 15.3 Intravenous Adenosine for Paroxysmal SVT
Adverse Effects
The adverse effects of adenosine are short-lived because of the drug’s ultra-short duration of action. The most frequent adverse effect is post-conversion bradycardia, including various degrees of AV block. The AV block is refractory to atropine, but resolves spontaneously within 60 seconds (26). Dipyridamole enhances the AV block produced by adenosine (26). Adenosine is contraindicated in patients with asthma because of reports of adenosine-induced bronchospasm (28), but more recent studies indicate that adenosine produces a sense of dyspnea, but not bronchospasm, in asthmatic subjects (29).
Refractory Tachycardias
Methylxanthines like theophylline block adenosine receptors and reduce the effectiveness of adenosine in terminating re-entrant tachycardias (26). This interaction has been minimized by the diminished use of theophylline as a bronchodilator. When PSVT does not respond to adenosine, the calcium channel blockers diltiazem or verapamil can be effective. (The dosing regimen for diltiazem is shown in Table 15.1, and the dosing regimen for verapamil was presented in the section on paroxysmal SVTs).
VENTRICULAR TACHYCARDIA
Ventricular tachycardia (VT) is a wide-QRS-complex tachycardia that has an abrupt onset, a regular rhythm, and a rate above 100 bpm (usually 140–200 bpm). The appearance can be monomorphic (uniform QRS complexes) or polymorphic (multiple QRS morphologies). VT rarely occurs in the absence of structural heart disease (30), and when it is sustained (i.e., lasts longer than 30 seconds), it can be an immediate threat to life.
VT versus SVT
Monomorphic VT can be difficult to distinguish from an SVT with prolonged AV conduction. This is demonstrated in Figure 15.5. The tracing in the upper panel shows a wide-QRS-complex tachycardia that looks like monomorphic VT. The tracing in the lower panel shows spontaneous conversion to sinus rhythm. Note that the QRS complex remains unchanged after the arrhythmia is terminated, revealing an underlying bundle branch block. Thus, the apparent VT in the upper panel is actually an SVT with a preexisting bundle branch block.
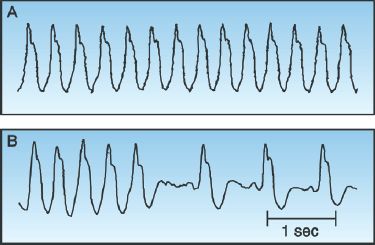
FIGURE 15.5 Upper panel shows a wide-QRS-complex tachycardia that looks like monomorphic VT. However, the lower panel shows spontaneous conversion to a sinus rhythm, which reveals an underlying bundle branch block, indicating that the rhythm in the upper panel is an SVT with a preexisting bundle branch block. Tracings courtesy of Richard M. Greenberg, M.D.
Clues
There are two abnormalities on an ECG that will identify VT as the cause of a wide-QRS-complex tachycardia.
1. The atria and ventricles beat independently in VT, and this results in AV dissociation on the ECG, where there is no fixed relationship between P waves and QRS complexes. This may not be evident on a single-lead tracing, and is more likely to be discovered on a 12-lead ECG. (P waves are most visible in the inferior limb leads and the anterior precordial leads.)
2. The presence of fusion beats like the one in Figure 15.6 is indirect evidence of VT. A fusion beat is produced by the retrograde transmission of a ventricular ectopic impulse that collides with a supra-ventricular (e.g., sinus node) impulse. The result is a hybrid QRS complex that is a mixture of the normal QRS complex and the ventricular ectopic impulse. The presence of a fusion beat (which should be evident on a single-lead ECG tracing) is indirect evidence of ventricular ectopic activity.
If there is no definitive evidence of VT on the ECG, the presence or ab-sence of heart disease can be useful; i.e., VT is the cause of 95% of wide-QRS-complex tachycardias in patients with underlying heart disease (31). Therefore, a wide-QRS-complex tachycardia should be treated as probable VT in any patient with underlying heart disease.
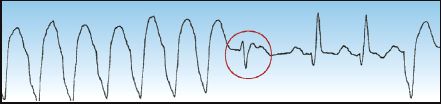
FIGURE 15.6 An example of a fusion beat (circled in red), which is a hybrid QRS complex produced by the collision of a ventricular ectopic impulse and a supraventricular (e.g., sinus node) impulse. The presence of fusion beats is evidence of ventricular ectopic activity.
Management
The management of patients with a wide-QRS-complex tachycardia can proceed as follows. This approach is organized in a flow diagram in Figure 15.7.
1. If there is evidence of hemodynamic compromise, electrical cardioversion is the appropriate intervention, regardless of whether the rhythm is VT or SVT with aberrant conduction. The shocks should be synchronized (timed with the QRS complex), with an initial shock of 100 J (biphasic or monophasic shocks) (30). This should terminate most cases of monomorphic VT, but shocks of 200 J (biphasic shocks) and up to 360 J (monophasic shocks) may be necessary.
2. If there is no hemodynamic compromise and the diagnosis of VT is certain, intravenous amiodarone should be used to terminate the arrhythmia. Amiodarone is the favored drug for suppression of monomorphic VT (4).
3. If there is no hemodynamic compromise and the diagnosis of VT is uncertain, the response to adenosine can be helpful because adenosine will abruptly terminate most cases of paroxysmal SVT, but will not terminate VT. If a wide-QRS-complex tachycardia is refractory to adenosine, the likely diagnosis is VT, and intravenous amiodarone is indicated for arrhythmia suppression.
Torsade de Pointes
Torsade de pointes (“twisting around the points”) is a polymorphic VT with QRS complexes that appear to be twisting around the isoelectric line of the ECG, as shown in Figure 15.8. This arrhythmia is associated with a prolonged QT interval, and it can be congenital or acquired. The acquired form is much more prevalent, and is caused by a variety of drugs and electrolyte abnormalities that prolong the QT interval (32,33). There is also a polymorphic VT that is associated with a normal QT interval, and the predisposing condition in this arrhythmia is myocardial ischemia (4).
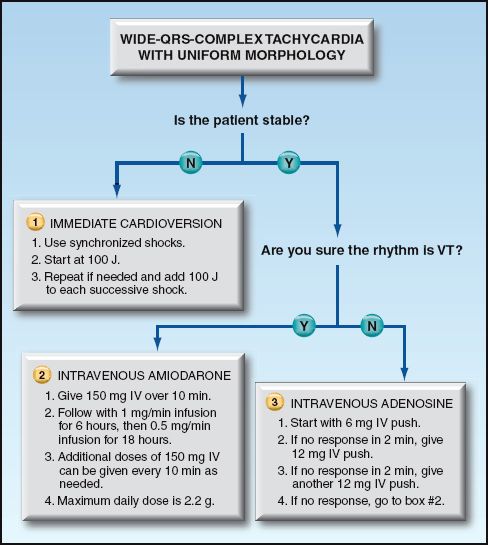
FIGURE 15.7 Flow diagram for the acute management of patients with wide-QRS-complex tachycardia. Based on recommendations in Reference 4.
Predisposing Factors
The drugs most frequently implicated in torsade de pointes are listed in Table 15.4 (33). The prominent offenders are antiarrhythmic agents (Class IA and III), macrolide antibiotics, neuroleptic agents, cisapride (a pro-motility drug), and methadone. The electrolyte disorders that prolong the QT interval include hypokalemia, hypocalcemia, and hypomagnesemia.
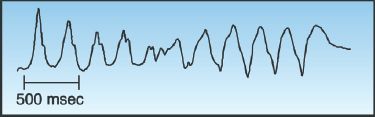
FIGURE 15.8 Torsade de pointes, a polymorphic ventricular tachycardia described as “twisting around the (isoelectric) points.” Tracing courtesy of Richard M. Greenberg, M.D.
Table 15.4 Drugs That Can Induce Torsade de Pointes
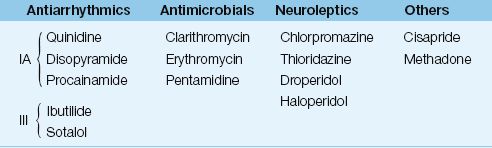
From Reference 33. For a complete list of drugs, go to www.torsades.org.
Measuring the QT Interval:
The QT interval is the ECG manifestation of ventricular depolarization and repolarization, and is measured from the onset of the QRS complex to the end of the T wave. The longest QT intervals are usually in precordial leads V3 and V4, and these leads are most reliable for assessing QT prolongation. The QT interval varies inversely with heart rate (e.g., an increase in heart rate shortens the QT interval), and therefore a rate-corrected QT interval (QTc) provides a more accurate assessment of QT prolongation. The accepted method for determining the QTc is to divide the QT interval by the square root of the R-R interval (33-35); i.e.,
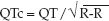
A normal QT interval corresponds to a QTc ″0.44 seconds, and a QTc >0.5 seconds represents an increased risk of torsade de pointes (35). The risk of torsade de points when the QTc = 0.45–0.5 seconds is not clear (33,35). There is no consensus on how to measure the QTc in highly irregular rhythms like atrial fibrillation.
Management
The management of polymorphic VT is summarized as follows:
1. Sustained polymorphic VT requires nonsynchronized electric cardio-version (i.e., defibrillation) (4).
2. For torsade de pointes, intravenous magnesium is used for pharmacological management (and can be combined with electrical cardioversion). There is no universal dosing regimen for magnesium in this setting. The ACLS guidelines recommend 1–2 grams magnesium sulfate (MgSO4) IV over 15 minutes (4), while another recommended regimen is 2 grams MgSO4 as an IV bolus, followed by a continuous magnesium infusion at 2–4 mg/min (33). Aggressive magnesium loading has no reported adverse consequences (even in patients with renal insufficiency), so the more aggressive magnesium regimen (33) should be the favored one.
3. Other measures for torsade de pointes include correcting high-risk electrolyte abnormalities and discontinuing high-risk drugs, to prevent recurrence.
4. For polymorphic VT with a normal QT interval, amiodarone or β-blockers (for myocardial ischemia) can help to prevent recurrences (4).
A FINAL WORD
The tachyarrhythmia that will dominate your time more than all the others combined is atrial fibrillation (AF). This is rarely a primary problem (in a non-coronary ICU), but it can require more attention than the primary problem. This arrhythmia is like an annoying person you meet who demands a lot of attention but is not threatening (not life-threatening, in the case of the arrhythmia).
The tachyarrhythmias that are an immediate threat to life (i.e., ventricular tachycardia) are not prevalent in non-coronary ICUs, even in patients with circulatory shock and multiorgan failure. In fact, in patients who are near death and are not resuscitated, the progression to ventricular asystole is usually populated by bradyarrhythmias (e.g. AV blocks). This paucity of troublesome ventricular tachycardia outside the coronary care unit may be explained by the trigger for ventricular tachycardia (i.e., focal, rather than global, myocardial ischemia), which is not a common occurrence in critical illness when coronary artery disease is not the dominant problem.
REFERENCES
Clinical Practice Guidelines
1. Fuster V, Ryden LE, Cannom DS, et al. 2011 ACCF/AHA/ESC focused updates incorporated into the ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation. J Am Coll Cardiol 2011; 57:e101–e198.
2. You JJ, Singer DE, Howard PA, et al. Antithrombotic therapy for atrial fibrillation. Antithrombotic Therapy and Prevention of Thrombosis, 9th ed. American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(Suppl):e531S–e575S.
3. The Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Guidelines for the management of atrial fibrillation. Europace 2010; 12:1360–1420.
4. Neumar RW, Otto CW, Link MS, et al. Part 8: adult advanced cardiovascular life support. 2010 American Heart Association Guidelines for Cardiopul-monary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122(Suppl):S729–S767.
Atrial Fibrillation
5. Mayson SE, Greenspon AJ, Adams S, et al. The changing face of postoperative atrial fibrillation: a review of current medical therapy. Cardiol Rev 2007; 15:231–241.
6. Davis EM, Packard KA, Hilleman DE. Pharmacologic prophylaxis of postoperative atrial fibrillation in patients undergoing cardiac surgery: beyond beta blockers. Pharmacotherapy 2010; 30:274e–318e.
7. Ozaydin M, Peker O, Erdogan D, et al. N-acetylcysteine for the prevention of postoperative atrial fibrillation: a prospective, randomized, placebo-controlled pilot study. Eur Heart J 2008; 29:625–631.
8. Guyton AC. The relationship of cardiac output and arterial pressure control. Circulation 1981; 64:1079–1088.
9. Siu C-W, Lau C-P, Lee W-L, et al. Intravenous diltiazem is superior to intravenous amiodarone or digoxin for achieving ventricular rate control in patients with acute uncomplicated atrial fibrillation. Crit Care Med 2009; 37:2174–2179.
10. Goldenberg IF, Lewis WR, Dias VC, et al. Intravenous diltiazem for the treatment of patients with atrial fibrillation or flutter and moderate to severe congestive heart failure. Am J Cardiol 1994; 74:884–889.
11. Olshansky B, RosenfeldLE, Warner AI, et al. The Atrial Fibrillation Follow-up Investigation of Rhythm Management (AFFIRM) study: approaches to control rate in atrial fibrillation. J Am Coll Cardiol 2004; 43:1201–1208.
12. Gray RJ. Managing critically ill patients with esmolol. An ultra-short-acting α-adrenergic blocker. Chest 1988;93:398–404.
13. Karth GD, Geppert A, Neunteufl T, et al. Amiodarone versus diltiazem for rate control in critically ill patients with atrial tachyarrhythmias. Crit Care Med 2001; 29:1149–1153.
14. Khan IA, Mehta NJ, Gowda RM. Amiodarone for pharmacological cardioversion of recent-onset atrial fibrillation. Int J Cardiol 2003; 89:239–248.
15. VerNooy RA, Mounsey P. Antiarrhythmic drug therapy in atrial fibrillation. Cardiol Clin 2004; 22:21–34.
16. Chow MSS. Intravenous amiodarone: pharmacology, pharmacokinetics, and clinical use. Ann Pharmacother 1996; 30:637–643.
17. Diltiazem hydrochloride. In: McEvoy GK (ed). AHFS Drug Information: 2012. Bethesda, MD: American Society of Health System Pharmacists, 2012:1961-1969.
18. Danias PG, Caulfield TA, Weigner MJ, et al. Likelihood of spontaneous conversion of atrial fibrillation to sinus rhythm. J Am Coll Cardiol 1998; 31:588–592.
19. Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
Multifocal Atrial Tachycardia
20. Kastor J. Multifocal atrial tachycardia. N Engl J Med 1990; 322:1713–1720.
21. Iseri LT, Fairshter RD, Hardeman JL, Brodsky MA. Magnesium and potassium therapy in multifocal atrial tachycardia. Am Heart J 1985; 312:21–26.
22. McLean RM. Magnesium and its therapeutic uses: a review. Am J Med 1994; 96:63–76.
Paroxysmal Supraventricular Tachycardias
23. Trohman RG. Supraventricular tachycardia: implications for the internist. Crit Care Med 2000; 28(Suppl):N129–N135.
24. Lim SH, Anantharaman V, Teo WS, et al. Comparison of treatment of supraventricular tachycardia by Valsalva maneuver and carotid sinus massage. Ann Emerg Med 1998; 31:30–35.
25. Rankin AC, Brooks R, Ruskin JM, McGovern BA. Adenosine and the treatment of supraventricular tachycardia. Am J Med 1992; 92:655–664.
26. Chronister C. Clinical management of supraventricular tachycardia with adenosine. Am J Crit Care 1993; 2:41–47.
27. McCollam PL, Uber W, Van Bakel AB. Adenosine-related ventricular asystole. Ann Intern Med 1993; 118:315–316.
28. Cushley MJ, Tattersfield AE, Holgate ST. Adenosine-induced bronchoconstriction in asthma. Am Rev Respir Dis 1984; 129:380–384.
29. Burki NK, Alam M, Lee L-Y. The pulmonary effects of intravenous adenosine in asthmatic subjects. Respir Res 2006; 7:139-146.
Ventricular Tachycardia
30. Gupta AK, Thakur RK. Wide QRS complex tachycardias. Med Clin N Am 2001; 85:245–266.
31. Akhtar M, Shenasa M, Jazayeri M, et al. Wide QRS complex tachycardia. Ann Intern Med 1988; 109:905–912.
32. Vukmir RB. Torsades de pointes: a review. Am J Emerg Med 1991; 9:250–262.
33. Gupta A, Lawrence AT, Krishnan K, et al. Current concepts in the mechanism and management of drug-induced QT prolongation and torsade de pointes. Am Heart J 2007; 153:891–899.
34. Sadanaga T, Sadanaga F, Yoo H, et al. An evaluation of ECG leads used to assess QT prolongation. Cardiology 2006; 105:149–154.
35. Al-Khatib SM, LaPointe NM, Kramer JM, Califf RM. What clinicians should know about the QT interval. JAMA 2003; 289:2120–2127.







Full access? Get Clinical Tree


