FIGURE 14.1 The sequence of chemical reactions involved in neutrophil activation, which generates a series of highly reactive oxygen metabolites that are stored in cytoplasmic granules. SOD = superoxide dismutase. See text for further explanation.
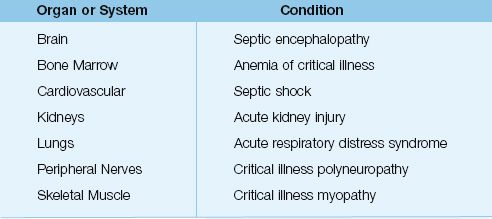
Table 14.1 Clinical Conditions Attributed to Inflammatory Injury
Clinical Syndromes
The following definitions have been adopted for the clinical syndromes associated with systemic inflammation (8,9):
1. The condition that is characterized by signs of systemic inflammation (e.g., fever, leukocytosis) is called the systemic inflammatory response syndrome (SIRS).
2. When SIRS is the result of an infection, the condition is called sepsis.
3. When sepsis is accompanied by dysfunction in one or more vital organs, or an elevated blood lactate level (>4 mM/L), the condition is called severe sepsis.
4. When severe sepsis is accompanied by hypotension that is refractory to volume infusion, the condition is called septic shock.
5. Inflammatory injury involving more than one vital organ is called multiorgan dysfunction syndrome (MODS), and the subsequent failure of more than one organ system is called (surprise!) multiorgan failure (MOF).
Systemic Inflammatory Response Syndrome (SIRS)
The diagnostic criteria for the SIRS are shown in Table 14.2. SIRS is a common condition; i.e., in one survey of patients in a surgical ICU, SIRS was identified in 93% of the patients (10). The presence of SIRS does not imply the presence of infection. Infection is identified in only 25 to 50% of patients with SIRS (10,11). The distinction between inflammation and infection is an essential ingredient in the rational approach to patients with fever and leukocytosis.
Table 14.2 Diagnostic Criteria for SIRS

Inflammatory Organ Failure
The organs most often damaged by systemic inflammation are the lungs, kidneys, cardiovascular system, and central nervous system (see Table 14.1). The most common manifestation of inflammatory organ injury is the acute respiratory distress syndrome (ARDS), which has been reported in 40% of patients with severe sepsis (12), and is one of the leading causes of acute respiratory failure in critically ill patients (see Chapter 23).
The number of organs that are damaged by inflammatory injury has important prognostic implications. This is shown in Figure 14.2, which includes surveys from the United States (12) and Europe (13) showing a direct relationship between the mortality rate and the number of organ failures related to inflammation. This demonstrates the lethal potential of uncontrolled systemic inflammation.
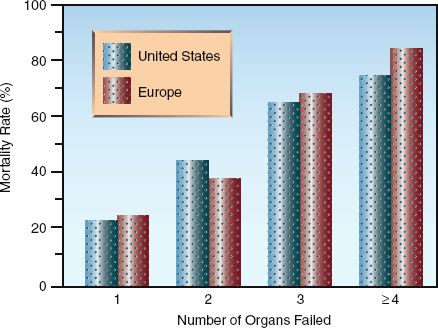
FIGURE 14.2 The relationship between mortality rate and the number of inflammation-related organ failures. Data from References 12 and 13.
SEPTIC SHOCK
Severe sepsis and septic shock (which are essentially the same condition with different blood pressures) have been implicated in one of every four deaths worldwide (9), and the incidence of these conditions is steadily rising. The mortality rate averages about 30–50% (12,14), and varies with age and the number of associated organ failures (as just described). The mortality rate is not related to the site of infection or the causative organism, including multidrug-resistant organisms (14). This observation is evidence that inflammation, not infection, is the principal determinant of outcome in severe sepsis and septic shock.
Hemodynamic Alterations
The hemodynamic alterations in septic shock are summarized below:
1. The principal hemodynamic problem is systemic vasodilatation (involving both arteries and veins), which reduces ventricular preload (cardiac filling pressures) and ventricular afterload (systemic vascular resistance). The vascular changes are attributed to the en-hanced production of nitric oxide (a free radical) in vascular endo-thelial cells (15).
2. Oxidant injury in the vascular endothelium (from neutrophil attachment and degranulation) leads to fluid extravasation and hypovolemia (15), which adds to the decreased ventricular filling from venodilatation.
3. Proinflammatory cytokines promote cardiac dysfunction (both systolic and diastolic dysfunction); however, the cardiac output is usually increased as a result of tachycardia and volume resuscitation (16).
4. Despite the increased cardiac output, splanchnic blood flow is typically reduced in septic shock (15). This can lead to disruption of the intestinal mucosa, thereby creating a risk for translocation of enteric pathogens and endotoxin across the bowel mucosa and into the systemic circulation (as described in Chapter 5). This, of course, will only aggravate the inciting condition.
The typical hemodynamic pattern in septic shock includes low cardiac filling pressures (CVP or wedge pressure), a high cardiac output (CO), and a low systemic vascular resistance (SVR); i.e.,
Typical Pattern: Low CVP / High CO / Low SVR
Because of the high cardiac output and peripheral vasodilatation, septic shock is also known as hyperdynamic shock or warm shock. In the advanced stages of septic shock, cardiac dysfunction is more prominent and the cardiac output is reduced, resulting in a hemodynamic pattern that resembles cardiogenic shock (i.e., high CVP, low CO, high SVR). A declining cardiac output in septic shock usually indicates a poor prognosis.
Tissue Oxygenation
As mentioned in Chapter 10, the impaired energy metabolism in septic shock is not the result of inadequate tissue oxygenation, but is caused by a defect in oxygen utilization in mitochondria (17,18). This condition is known as cytopathic hypoxia (17), and the culprit is oxidant-induced inhibition of cytochrome oxidase and other proteins in the electron transport chain (19). A decrease in oxygen utilization would explain the observation shown in Figure 14.3, where the PO2 in skeletal muscle is increased in patients with severe sepsis (19).
The proposed decrease in oxygen utilization in sepsis is not consistent with the increase in whole-body O2 consumption that is often observed in sepsis. This discrepancy can be resolved by proposing that the in-creased O2 consumption in sepsis is not a reflection of aerobic metabolism, but is a manifestation of the increased O2 consumption that occurs during neutrophil activation (i.e., the respiratory burst) (21).
Clinical Implications
The discovery that tissue oxygenation is (more than) adequate in severe sepsis and septic shock has important implications because it means that efforts to improve tissue oxygenation in these conditions (e.g., with blood transfusions) are not justified.
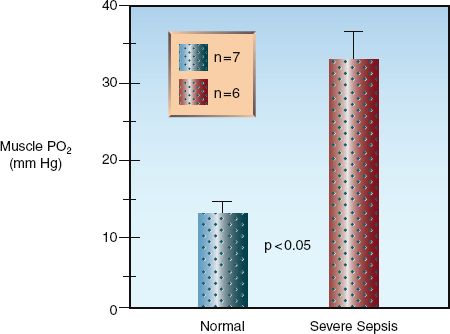
FIGURE 14.3 Direct measurements of tissue PO2 in the forearm muscles of healthy volunteers and patients with severe sepsis. The height of the columns represents the mean value for each group, and the crossbars represent the standard error of the mean. Data from Reference 20.
Serum Lactate Levels
As mentioned in Chapter 10, the increase in serum lactate levels in severe sepsis and septic shock is not the result of inadequate tissue oxygenation, but instead appears to be the result of enhanced production of pyruvate and inhibition of pyruvate dehydrogenase (22,23), the enzyme that converts pyruvate to acetyl coenzyme A in mitochondria. Endotoxin and other bacterial cell wall components have been implicated in the inhibition of this enzyme (22). This mechanism of lactate accumulation is consistent with the notion that tissue oxygenation is not impaired in severe sepsis and septic shock.
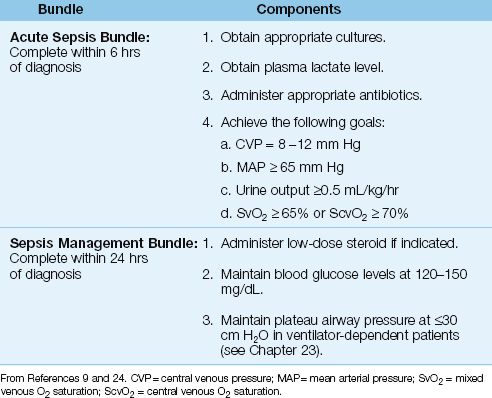
Table 14.3 The Management of Septic Shock Using Bundles
Management
The management of septic shock is outlined in Table 14.3, and is organized in “bundles,” which are sets of instructions that must be followed without deviation to provide a survival benefit. The bundles in Table 14.3 are from the “Surviving Sepsis Campaign” (an internationally recognized guideline for the management of septic shock) (9), and adherence to the instructions in these bundles has been shown to improve survival in patients with septic shock (24). The acute sepsis bundle is considered the most important, and must be completed within 6 hours after the diagnosis of septic shock.
Volume Resuscitation
Volume resuscitation is often necessary in septic shock because cardiac filling pressures are reduced from venodilatation and fluid extravasation. The following recommendation for volume resuscitation is taken from the Surviving Sepsis Campaign guidelines (9), and it requires the insertion of a central venous catheter to monitor the central venous pressure (CVP).
1. Infuse 500–1,000 mL of crystalloid fluid or 300–500 mL of colloid fluid over 30 minutes.
2. Repeat as needed until the CVP reaches 8 mm Hg, or 12 mm Hg in ventilator-dependent patients.
THE CVP: The use of the CVP in the above protocol is problematic for two reasons. First, volume resuscitation will be delayed by the time required to insert the central line and obtain a chest x-ray to check for proper catheter placement. Secondly, the consensus opinion is that the CVP should NOT be used to guide fluid management because it is not an accurate reflection of the circulating blood volume. The discrepancy between the CVP and circulating blood volume is demonstrated in Figure 11.2 (Chapter 11). If CVP measurements are not available, a volume of at least 20 mL/kg (crystalloid fluid) can be used for the volume resuscitation (25).
After the initial period of volume resuscitation, the infusion rate of intravenous fluids should be reduced to avoid unnecessary fluid accumulation. A positive fluid balance is associated with increased mortality in septic shock (26), so attention to avoiding fluid accumulation will im-prove the chances of a favorable outcome.
Vasopressors
If hypotension persists after the initial volume resuscitation, infusion of a vasoconstrictor drug (vasopressor) like norepinephrine or dopamine should begin (9). Vasoconstrictor drugs must be infused through a central venous catheter, and the goal is to achieve a mean arterial pressure (MAP) ≥65 mm Hg (9).
1. For norepinephrine, start with a dose rate of 0.1 ∝g/kg/min and titrate upward as needed. Dose rates up to 3.3 ∝g/kg/min are successful in raising the blood pressure in a majority of patients with septic shock (27). If the desired MAP is not achieved at a dose rate of 3–3.5 ∝g/kg/min, add dopamine as a second vasopressor.
2. For dopamine, start at a dose rate of 5 ∝g/kg/min and titrate up-ward as needed. Vasoconstriction is the predominant effect at dose rates above 10 ∝g/kg/min (27). If the desired MAP is not achieved with a dose rate of 20 ∝g/kg/min, add norepinephrine as a second vasopressor.
Norepinephrine is favored by many because it is more likely to raise the blood pressure than dopamine, and is less likely to promote arrhythmias (27). However, neither agent has proven superior to the other for improving the outcome in septic shock (26). (Norepinephrine and dopamine are described in more detail in Chapter 53.)
VASOPRESSIN: When hypotension is refractory to norepinephrine and dopamine, vasopressin may be effective in raising the blood pressure. (Vasopressin is used as an additional pressor rather than a replacement for norepinephrine or dopamine.) The dose range for vasopressin is 0.01–0.04 units/min, but the popular dose rate in septic shock is 0.03 units/min (9). Vasopressin is a pure vasoconstrictor that can promote splanchnic and digital ischemia, especially at high dose rates. Although vasopressin may help in raising the blood pressure, the accumulated experience with vasopressin shows no influence on outcomes in septic shock (28).
Corticosteroids
Corticosteroids have two actions that are potentially beneficial in septic shock: they have antiinflammatory activity, and they magnify the vasoconstrictor response to catecholamines. Unfortunately, after more than 50 years of investigations, there is no convincing evidence that steroids provide any benefit in the treatment of septic shock (29,30). Yet steroid therapy continues to be popular in septic shock. The following comments reflect the current recommendations regarding steroid therapy in septic shock (9).
1. Steroid therapy should be considered in cases of septic shock where the blood pressure is poorly responsive to intravenous fluids and vasopressor therapy. Evidence of adrenal insufficiency (by the rapid ACTH stimulation test) is not required.
2. Intravenous hydrocortisone is preferred to dexamethasone (because of the mineralocorticoid effects of hydrocortisone), and the dose should not exceed 300 mg daily (to limit the risk of infection).
3. Steroid therapy should be continued as long as vasopressor therapy is required.
Despite the persistent use of steroids in septic shock, it seems that if a drug effect is not apparent after 50 years of investigation (!), then it’s time to conclude that the drug does not produce the effect.
Antimicrobial Therapy
Delays in initiating appropriate antibiotic therapy are associated with an increased mortality rate in severe sepsis and septic shock (31), and this has prompted the recommendation that antibiotic therapy should be started within one hour of the diagnosis of severe sepsis or septic shock (9). This leaves little time to identify potential pathogens, so the initial antibiotic(s) should have a broad spectrum of activity. See Chapter 43 for recommendations concerning empiric antibiotic coverage for patients with suspected sepsis.
BLOOD CULTURES: One dose of an intravenous antibiotic can sterilize blood cultures within a few hours, so blood cultures should be obtained prior to administering antibiotics. At least 2 sets of blood cultures are recommended (9). Two sets of blood cultures will detect about 90% of bloodstream infections, while 3 sets of blood cultures will detect close to 98% of bloodstream infections (32). The yield from blood cultures is influenced by the volume of blood that is cultured, and a volume of at least 20 mL is recommended for each set of blood cultures (33).
Table 14.4 Clinical Manifestations of Anaphylaxis
ANAPHYLAXIS
Anaphylaxis is an acute multiorgan dysfunction syndrome produced by the immunogenic release of inflammatory mediators from basophils and mast cells. The characteristic feature is an exaggerated immunoglobulin E (IgE) response to an external antigen; i.e., a hypersensitivity reaction. The manifestations of anaphylaxis typically involve the skin, lungs, gastrointestinal tract, and cardiovascular system (35). Identical manifestations can occur without the involvement of IgE; these are called anaphylactoid reactions, and are not immunogenic in origin (36). Common triggers for anaphylactic reactions include food, antimicrobial agents, and insect bites, while common triggers for anaphylactoid reactions include opiates and radiocontrast dyes. Anaphylaxis can also appear without an identifiable external trigger.
Clinical Features
Anaphylactic reactions are typically abrupt in onset, and appear within minutes of exposure to the external trigger. Some reactions are delayed, and can appear as late as 72 hours after exposure (35). A characteristic feature of anaphylactic reactions is edema and swelling in the involved organ, caused by increased vascular permeability with extravasation of fluid. As much as 35% of the intravascular volume can be lost within 10 minutes in severe anaphylactic reactions (35).
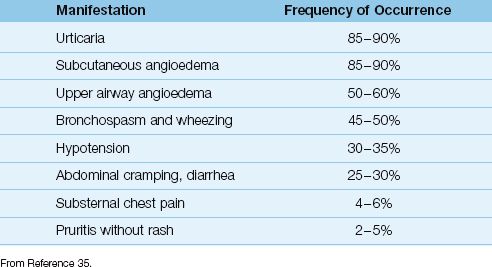
The clinical manifestations of anaphylaxis are shown in Table 14.4, and are listed by their frequency of occurrence. The most common manifestations are urticaria and subcutaneous angioedema (typically involving the face), and the most concerning manifestations are angioedema of the upper airway (e.g., laryngeal edema), bronchospasm, and hypotension. The most feared manifestation of anaphylaxis is profound hypotension with evidence of systemic hypoperfusion, which represents anaphylactic shock.
Management
The management of anaphylaxis includes drugs that halt the progress of anaphylactic reactions (i.e., epinephrine), and drugs that alleviate signs and symptoms (e.g., bronchodilators).
Epinephrine
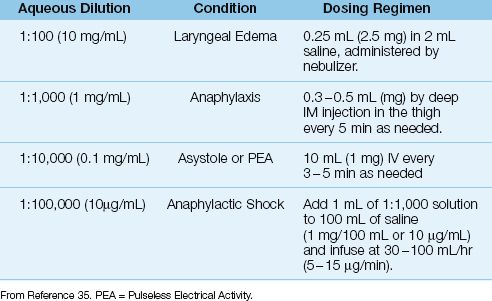
Epinephrine is the most effective drug available for treating anaphylaxis, and is capable of blocking the release of inflammatory mediators from sensitized basophils and mast cells. The drug is available in a (confusing) variety of aqueous solutions, and these are shown in Table 14.5. The usual treatment for anaphylactic reactions is 0.3–0.5 mg of epinephrine (0.3–0.5 mL of 1:1000 epinephrine solution) administered by deep intramuscular (IM) injection in the lateral thigh, and repeated every 5 minutes if necessary (35). Drug absorption is slower with subcutaneous injection (36), and with drug injection in the deltoid muscle instead of the thigh (35). Epinephrine can be nebulized for patients with laryngeal edema using the dosing regimen shown in Table 14.5; however, the efficacy of nebulized epinephrine is unclear.
GLUCAGON: The actions of epinephrine to inhibit degranulation of mast cells and basophils is mediated by β-adrenergic receptors, and ongoing therapy with β-receptor antagonists can attenuate or eliminate the response to epinephrine. When anaphylactic reactions are refractory to epinephrine in patients receiving β-blocker drugs, glucagon can be effective (for reasons described in Chapter 54). The dose of glucagon is 1–5 mg by slow intravenous injection (over 5 min), followed by a continuous infusion at 5–15 µg/min, titrated to the desired response (35). Glucagon can trigger vomiting, and patients with depressed consciousness should be placed on their side to limit the risk of aspiration when glucagon is administered.
Second-Line Agents
The following drugs can be given after epinephrine is administered, and should never be used as a replacement for epinephrine.
ANTIHISTAMINES: Histamine receptor antagonists are often used for cutaneous anaphylactic reactions, and can help in alleviating pruritis. The histamine H1 blocker diphenhydramine (25–50 mg PO, IM, or IV) and the histamine-H2 blocker ranitidine (50 mg IV or 150 mg PO) should be given together because they are more effective in combination.
Table 14.5 Aqueous Epinephrine Solutions and Their Clinical Uses
ANTIHISTAMINES: Histamine receptor antagonists are often used for cutaneous anaphylactic reactions, and can help in alleviating pruritis. The histamine H1 blocker diphenhydramine (25–50 mg PO, IM, or IV) and the histamine-H2 blocker ranitidine (50 mg IV or 150 mg PO) should be given together because they are more effective in combination.
BRONCHODILATORS: Inhaled β2-receptor agonists like albuterol are used to relieve bronchospasm, and are administered by nebulizer (2.5 mL or a 0.5% solution) or by metered-dose inhaler.
CORTICOSTEROIDS: Despite the popularity of steroids for treating hypersensitivity reactions, there is no evidence that steroids are effective in reversing, slowing, or preventing the recurrence of anaphylactic reactions (35). As a result, the most recent practice guideline on treating anaphylaxis does not include a recommendation for steroid therapy (35).
Anaphylactic Shock
Anaphylactic shock is an immediate threat to life, with profound hypotension from systemic vasodilatation and massive fluid loss through leaky capillaries (35). The hemodynamic alterations in anaphylactic shock are similar to those in septic shock, but are often more pronounced. Because of the potential for rapid deterioration, anaphylactic shock requires prompt and aggressive management using the measures described next.
Epinephrine
There is no standardized dosing regimen for epinephrine in anaphylactic shock, but the epinephrine infusion regimen in Table 14.5, which uses dose rates of 5–15 µg/min, has been cited for its efficacy (35). An intravenous bolus dose of epinephrine (5–10 µg) can precede the continuous drug infusion (37).
Volume Resuscitation
Aggressive volume resuscitation is essential in anaphylactic shock be-cause at least 35% of the intravascular volume can be lost through leaky capillaries (35), which is enough to produce hypovolemic shock (see Chapter 11). Volume resuscitation can begin by infusing 1–2 liters of crystalloid fluid (or 20 mL/kg), or 500 mL of iso-oncotic colloid fluid (e.g., 5% albumin), over the first 5 minutes (35). Thereafter, the infusion rate of fluids should be tailored to the clinical condition of the patient.
Refractory Hypotension
Persistent hypotension despite epinephrine infusion and volume resuscitation can be managed by adding glucagon or another vasopressor such as norepinephrine or dopamine (the dosing regimens for these drugs have been described earlier).
A FINAL WORD
Another Look at Inflammatory Injury
The discovery that inflammation is the source of multiorgan failure and fatal outcomes in septic shock has created interest in therapies aimed at inhibiting the inflammatory response in septic shock. So far, these therapies have failed to produce the anticipated benefits. This is not unexpected, because the problem with inflammatory injury is not the inflammation, but the inability of the host to protect itself from inflammatory injury. Since the damage inflicted by inflammation is largely due to oxidation (i.e., oxidant cell injury), inflammatory injury is a manifestation of oxidant stress, where the production of oxidants (like the reactive oxygen metabolites in Figure 14.1) overwhelms the body’s endogenous antioxidant defenses. Therefore, inflammatory injury can be the result of inadequate antioxidant protection.
Conditions like severe sepsis and septic shock create an oxidation-rich environment in tissues, which requires an antioxidant-rich defense system. However, antioxidant support is never provided for critically ill patients. It seems likely that persistent oxidation will eventually deplete endogenous antioxidants like glutathione (the major intracellular antioxidant) and vitamin E (which protects cell membranes from oxidant injury), ensuring the progressive march of inflammatory injury and multiorgan failure. There is some evidence that daily administration of endogenous antioxidants improves outcomes in septic shock (38), and the promise of this approach deserves much more attention.
REFERENCES
Moore W. The Knife Man: Blood, Body Snatching, and the Birth of Modern Surgery. New York: Broadway Books, 2005.
Inflammatory Injury
1. Pinsky MR, Matuschak GM. Multiple systems organ failure: failure of host defense mechanisms. Crit Care Clin 1989; 5:199–220.
2. Pinsky MR, Vincent J-L, Deviere J, et al. Serum cytokine levels in human septic shock: Relation to multiple-system organ failure and mortality. Chest 1993; 103:565–575.
3. Fujishima S, Aikawa N. Neutrophil-mediated tissue injury and its modulation. Intensive Care Med 1995; 21:277–285.
4. Babior BM. The respiratory burst of phagocytes. J Clin Invest 1984; 73:599–601.
5. Bernovsky C. Nucleotide chloramines and neutrophil-mediated cytotoxicity. FASEB Journal 1991; 5:295–300.
6. Halliwell B, Gutteridge JMC. The chemistry of free radicals and related ‘reactive species’. In: Free Radicals in Biology and Medicine. 4th ed. New York: Oxford University Press, 2007:30–79.
7. Niki E, Yamamoto Y, Komura E, Sato K. Membrane damage due to lipid oxidation. Am J Clin Nutr 1991; 53:201S–205S.
8. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference Committee. Definitions of sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992; 101:1644–1655.
9. Dellinger RP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock. Intensive Care Med 2008; 34:17–60.
10. Pittet D, Rangel-Frausto S, Li N, et al. Systemic inflammatory response syndrome, sepsis, severe sepsis, and septic shock: incidence, morbidities and outcomes in surgical ICU patients. Intensive Care Med 1995; 21:302–309.
11. Rangel-Frausto MS, Pittet D, Costigan M, et al. Natural history of the systemic inflammatory response syndrome (SIRS). JAMA 1995; 273:117–123.
12. Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29:1303–1310.
13. Vincent J-L, de Mendonca A, Cantraine F, et al. Use of the SOFA score to assess the incidence of organ dysfunction/failure in intensive care units: Results of a multicenter, prospective study. Crit Care Med 1998; 26:1793–1800.
Severe Sepsis and Septic Shock
14. Zahar J-R, Timsit J-F, Garrouste-Orgeas M, et al. Outcomes in severe sepsis and patients with septic shock: pathogen species and infection sites are not associated with mortality. Crit Care Med 2011; 39:1886–1895.
15. Abraham E, Singer M. Mechanisms of sepsis-induced organ dysfunction. Crit Care Med 2007; 35:2409–2416.
16. Snell RJ, Parillo JE. Cardiovascular dysfunction in septic shock. Chest 1991; 99:1000–1009.
17. Fink MP. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin 2001; 17:219–237.
18. Ruggieri AJ, Levy RJ, Deutschman CS. Mitochondrial dysfunction and resuscitation in sepsis. Crit Care Clin 2010; 26:567–575.
19. Muravchick S, Levy RJ. Clinical implications of mitochondrial dysfunction. Anesthesiology 2006; 105:819–837.
20. Sair M, Etherington PJ, Winlove CP, Evans TW. Tissue oxygenation and perfusion in patients with systemic sepsis. Crit Care Med 2001; 29:1343–1349.
21. Vlessis AA, Goldman RK, Trunkey DD. New concepts in the pathophysiology of oxygen metabolism during sepsis. Br J Surg 1995; 82:870–876.
22. Thomas GW, Mains CW, Slone DS, et al. Potential dysregulation of the pyruvate dehydrogenase complex by bacterial toxins and insulin. J Trauma 2009; 67:628–633.
23. Loiacono LA, Shapiro DS. Detection of hypoxia at the cellular level. Crit Care Clin 2010; 26:409–421.
24. Barochia AV, Cui X, Vitberg D, et al. Bundled care for septic shock: an analysis of clinical trials. Crit Care Med 2010; 38:668–678.
25. The Surviving Sepsis Campaign website (www.survivingsepsis.org); accessed Sept 15, 2012.
26. Boyd JH, Forbes J, Nakada T-a, et al. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
27. Hollenberg SM. Inotropes and vasopressor therapy of septic shock. Crit Care Clin 2009; 25:781–802.
28. Polito A, Parisini E, Ricci Z, et al. Vasopressin for treatment of vasodilatory shock: an ESICM systematic review and meta-analysis. Intensive Care Med 2012; 38:9–19.
29. Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med 2008; 358:111–124.
30. Sherwin RL, Garcia AJ, Bilkovski R. Do low-dose corticosteroids improve mortality or shock reversal in patients with septic shock? J Emerg Med 2012; 43:7–12.
31. Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med 2010; 38:1045–1053.
32. Lee A, Mirrett S, Reller B, Weinstein MP. Detection of bloodstream infections in adults: how many blood cultures are needed? J Clin Microbiol 2007; 45:3546–3548.
33. Cockerill FR III, Wilson JW, Vetter EA, et al. Optimal testing parameters for blood cultures. Clin Infect Dis 2004; 38:1724–1730.
34. Marik PE, Preiser J-C. Toward understanding tight glycemic control in the ICU. Chest 2010; 137:544–551.
Anaphylaxis
35. Lieberman P, Nicklas RA, Oppenheimer J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol 2010; 126:480.e1–480.e42.
36. Simons FER, Gu X, Simons KJ. Epinephrine absorption in adults: intramuscular versus subcutaneous injection. J Allergy Clin Immunol 2001; 108(5):871–873.
37. Sampson HA, Munoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report – second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. Ann Emerg Med 2006; 47:373–380.
A Final Word
38. Angstwurm MWA, Engelmann L, Zimmermann T, et al. Selenium in intensive care (SIC): results of a prospective randomized placebo-controlled study in patients with severe systemic inflammatory response syndrome, sepsis, and septic shock. Crit Care Med 2007; 35:118–126.







Full access? Get Clinical Tree


