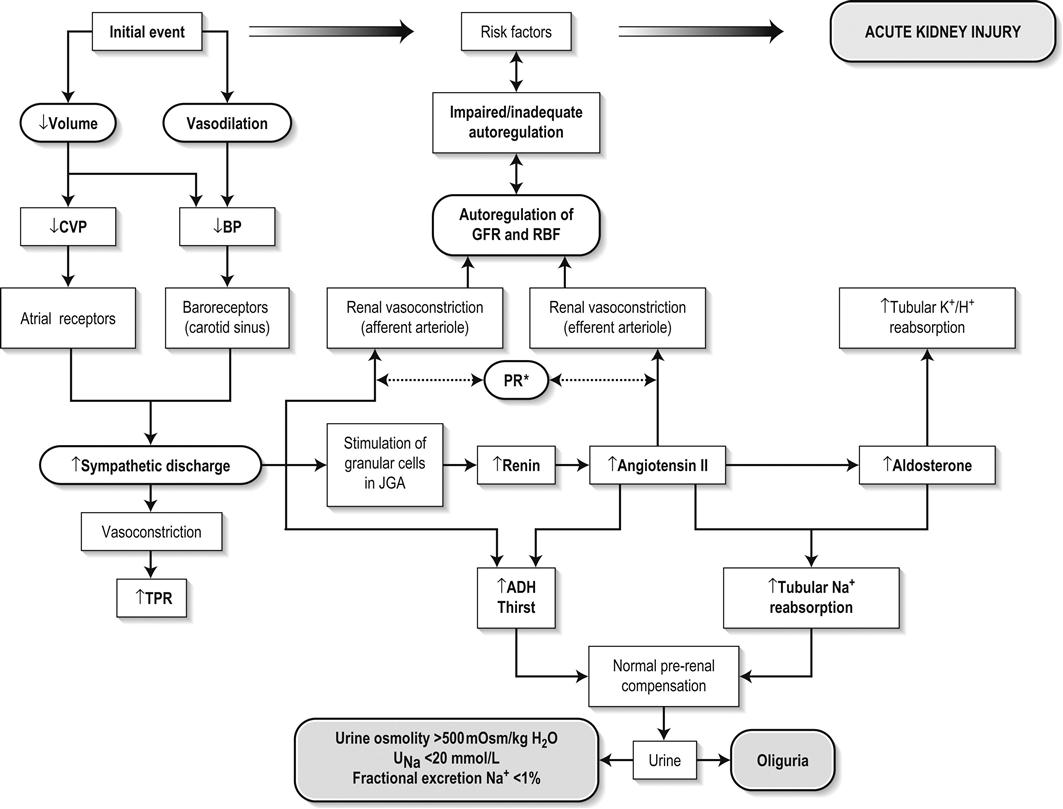
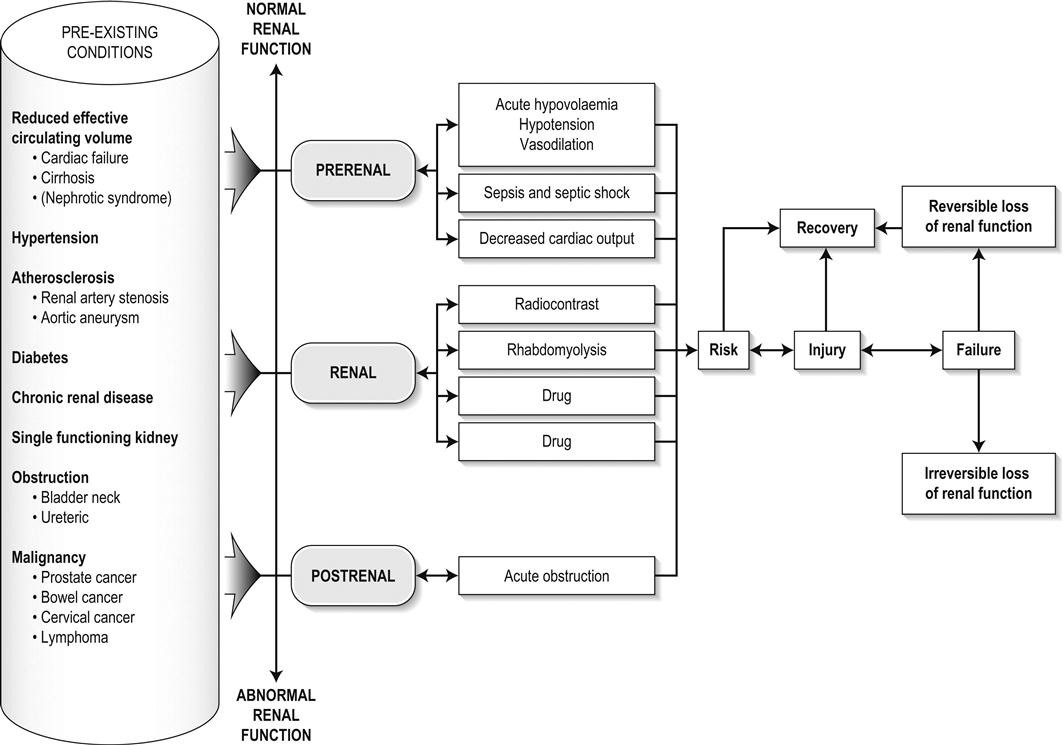
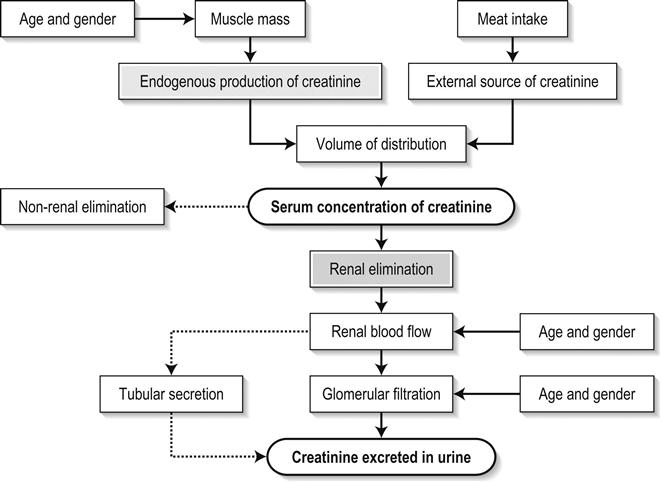
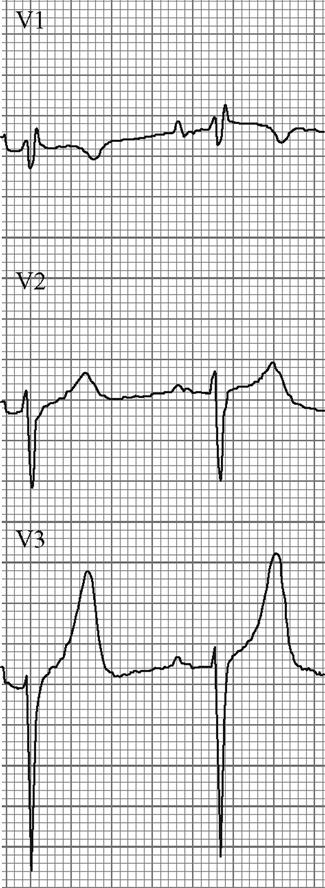
Edited by George Jelinek Nicholas Adams and Linas Dziukas The basic process in acute kidney injury (AKI) is a rapid (hours to days) reduction in the glomerular filtration rate (GFR) due to renal hypoperfusion, damage to glomeruli, tubules, interstitium or blood vessels, or obstruction to urine flow. The GFR is inversely related to the serum creatinine (SCr) concentration and the diagnosis of AKI is made when there is an acute increase in the SCr concentration, with or without a decrease in the urine output. A simple definition of AKI is an acute and sustained (lasting for 48 hours or more) increase in the SCr of 44 μmol/L if the baseline is less than 221 μmol/L, or an increase in the SCr of more than 20% if the baseline is more than 221 μmol/L. A more comprehensive definition (the RIFLE system) is used to classify persons with acute impairment of renal function (Table 10.1.1) [1]. Table 10.1.1 RIFLE classification of acute renal failure The causes of AKI are grouped according to the source of renal injury: prerenal (hypoperfusion), renal (parenchymal) and post-renal (obstructive). More than one cause can be present simultaneously. Prerenal AKI is initially an adaptive response to severe volume depletion and hypotension in structurally intact nephrons. Prerenal AKI that is prolonged or inadequately treated can be followed by parenchymal renal damage (acute tubular necrosis). Prerenal AKI is a potentially reversible cause of acute renal failure (ARF). Reductions in renal blood flow (RBF) and GFR occur in the setting(s) of hypovolaemia, hypotension, oedematous states with a reduced ‘effective’ circulating volume (cardiac failure, hepatic cirrhosis, nephrotic syndrome) or impaired renal perfusion (renal artery stenosis, hepatorenal syndrome). Drugs that interfere with renal autoregulation (e.g. prostaglandin inhibitors, angiotensin-converting enzyme [ACE] inhibitors or angiotensin II receptor antagonists) can reduce glomerular perfusion [2]. The physiological responses to volume depletion and hypotension, and the link to prerenal AKI are shown in Figure 10.1.1. Ischaemic, cytotoxic or inflammatory processes may damage the renal parenchyma. The causes of the damage can be grouped according to the major structures that are damaged: vessels, glomeruli, renal tubules or renal interstitial tissue. Vascular causes involving the larger vessels include acute thrombosis of the renal artery, embolism of the renal arteries, renal artery dissection and renal vein thrombosis. Microvascular causes include vasculitis, malignant hypertension and thrombotic microangiopathy. The glomeruli are the site of injury in acute glomerulonephritis which can cause proteinuria, haematuria, nephrotic syndrome or nephritic syndrome. A number of different forms of glomerulonephritis have been described, generally diagnosed by the histological changes seen on renal biopsy. The distinction between these forms is not of direct concern for the emergency practitioner. Acute tubular necrosis (ATN) is the most common pathological process that causes AKI. While the terminology suggests that the main cause is tubular damage, the actual pathophysiology is more complex: impaired autoregulation and marked intrarenal vasoconstriction (the main mechanism for the greatly reduced GFR), tubular damage (with cytoskeleton breakdown), increased tubuloglomerular feedback, endothelial cell injury, fibrin deposition in the microcirculation, release of cytokines, activation of inflammation and activation of the immune system [3]. ATN is often classified as ischaemic ATN or cytotoxic ATN but both processes may be present in some patients. Ischaemic ATN represents an advanced form of prerenal AKI, but the distinction between these two entities is based on histopathological changes and is of little use to the clinician. Important causes of cytotoxic ATN are listed in Table 10.1.2. Non-steroidal anti-inflammatory drugs (NSAIDs), ACE inhibitors and angiotensin receptor blockers (ARBs) often cause a gradual and asymptomatic decrease in the GFR, but can also cause AKI. NSAIDs do not impair renal function in healthy persons, but can reduce the GFR in the elderly with atherosclerotic cardiovascular disease, in persons with chronic renal failure, when chronic prerenal hypoperfusion is present (e.g. cardiac failure, cirrhosis) or in persons using diuretics and calcium channel blockers [4]. AKI may occur after the administration of intravenous or intra-arterial radiocontrast agents. A number of risk factors have been identified for this, the most important being pre-existing renal impairment, hypovolaemia, a large contrast load and the use of hyperosmolar contrast agents [5]. Drugs that alter angiotensin levels (ACE inhibitors and ARBs) reduce renal perfusion by their antihypertensive effects or by impairing vasoconstriction of the efferent arteriole when renal perfusion is reduced by renal artery stenosis. The nephrotoxicity of haem pigments (myoglobin and haemoglobin) is enhanced by volume depletion, low urine flow rates and possibly low urine pH. Table 10.1.2 Causes of toxic acute tubular necrosis Exogenous agents Radiocontrast Non-steroidal anti-inflammatory drugs Antibiotics: aminoglycosides, amphotericin B Antiviral drugs: acyclovir, foscarnet Immunosuppressive drugs: ciclosporin Organic solvents: ethylene glycol Poisons: snake venom, paraquat, paracetamol Chemotherapeutic drugs: cisplatin Herbal remedies Heavy metals Endogenous agents Haem pigments: haemoglobin, myoglobin Uric acid Myeloma proteins Correct intravascular volume depletion Maintain perfusion pressure Choice of resuscitation fluid Diuresis in rhabdomyolysis Avoid nephrotoxins Use derived GFR or creatinine clearance when calculating drug doses Abnormalities of renal interstitial structure and function are only one feature of ATN, but represent the primary abnormality in acute tubulointerstitial nephritis (ATIN). The damage in ATIN is due to immunological mechanisms, the most important involving cell-mediated immunity. ATIN is usually due to an allergic reaction to a drug, commonly antibiotics (β-lactam antibiotics, sulphonamides, fluoroquinolones), NSAIDs, cyclooxygenase-2 inhibitors, proton pump inhibitors, diuretics, phenytoin, carbamazepine and allopurinol. Obstructive uropathy refers to the functional or structural processes in the urinary tract that impede the normal flow of urine and obstructive nephropathy is the renal damage caused by the obstruction. Hydronephrosis and hydroureter refer to dilatation of the renal urinary collecting system and the ureters, respectively. They may occur in the absence of obstruction and, conversely, may be absent in some patients with obstruction. Casts or crystals within the renal tubular lumen can cause intrarenal obstruction. Extrarenal obstruction can develop in the urethra, bladder, ureter or the pelvi-ureteric junction. Obstructive uropathy in adults is commonly caused by prostate disease or retroperitoneal neoplasm (cancer of the cervix, uterus, bladder, ovary or colon). Metastatic cancer, lymphomas or inflammatory processes in the retroperitoneum (appendicitis, diverticulitis, Crohn’s disease) or a neurogenic bladder can also cause obstructive uropathy. Bilateral renal stones are an uncommon cause of obstructive uropathy. Obstructive nephropathy usually develops gradually and can cause chronic renal failure if the obstruction involves the urethra, the bladder or both ureters. Unilateral ureteric obstruction will cause AKI only if it involves a single functioning kidney. Studies of the pathogenesis of community-acquired ARF have produced conflicting results. In one study, the major processes were identified as prerenal in 70% of cases, renal in 11% of cases and post-renal in 17% of cases [6]. There are geographical differences in the causes of ATN. In Africa, India, Asia and Latin America, ATN is usually caused by infections (e.g. diarrhoeal illnesses, malaria, leptospirosis), ingestion of plants or medicinal herbs, envenomation, intravascular haemolysis due to glucose-6-phosphate dehydrogenase deficiency or poisoning. The rate and volume of intravenous fluid given to hypovolaemic persons depends on the nature of the intravascular depletion, the blood pressure and heart rate, the (estimated) volume of fluid lost, cardiac function and ongoing circulatory losses. The response to treatment is evaluated by simple bedside measurements (heart rate, blood pressure, urine output). Most studies on the prevention of ATN after rhabdomyolysis have been in persons with crush injury after earthquakes, where the incidence of AKI is about 50%. In this situation, fluid resuscitation should, if possible, begin before the crush is relieved. These patients may require massive amounts of fluid because of fluid sequestration in the injured muscles. The goal of intravenous fluid treatment is to produce a urine output of 200–300 mL/h while myoglobinuria (discoloured urine) persists. There is no evidence to support this rate of fluid replacement in persons who have rhabdomyolysis and AKI without crush injury, although a urine output of 100 mL/h would be reasonable while the urine is discoloured. The intravenous administration of mannitol and sodium bicarbonate to produce an alkaline diuresis as a means of preventing ATN in severe rhabdomyolysis has not been shown to be effective [7]. The incidence of radiocontrast nephropathy can be reduced by saline infusion to produce intravascular volume expansion and by using low osmolar contrast agents. N-acetyl cysteine administration before and after radiocontrast administration does not appear to be effective [5]. The diagnosis of AKI should be considered when there is a decrease in urine output or an elevated SCr concentration. The clinical features depend on the pre-existing conditions that increase the risk of developing AKI, the initiating factor(s) and the effects of AKI (Fig. 10.1.2). The history should include a detailed drug history, enquiry about recent invasive vascular or radiological procedures and any family history of renal disease. This is followed by clinical examination and evaluation of investigations. A number of key issues then need to be resolved (Table 10.1.3). Table 10.1.3 Evaluation of acute kidney injury Assess the intravascular volume Look for renovascular disease Look for symptoms or signs of obstruction to urine flow Systematic search for presence of infection or sepsis Evaluate for pre-existing renal disease or chronic renal failure Obtain a detailed history of medication or drug use Consider possibility of glomerulonephritis Imprecise terminology, such as ‘dry’ or ‘dehydrated’, should be avoided. ‘Dehydration’ refers to situations where more water than electrolyte(s) has been lost, shrinking body cells and increasing the serum sodium concentration and osmolality. In other words, ‘dehydration’ means water depletion. Hypovolaemia is a decrease in the intravascular volume due to loss of blood (haemorrhage, trauma) or loss of sodium and water (e.g. vomiting, diarrhoea, sequestration of fluid in the bowel, etc.). The (bedside) assessment of the (extracellular) volume status determines the initial resuscitation strategy. This involves evaluation of heart rate and blood pressure, the state of the skin and mucous membranes and the jugular venous pulse. The examination also includes auscultation of the lungs (for pulmonary crackles), abdominal examination (for ascites or masses) and examination of the legs (for peripheral oedema). The ‘typical’ features of intravascular volume depletion (tachycardia or hypotension or both, in the supine position, or postural hypotension) are not as consistent or reliable as implied by many textbook descriptions. The presence of (supine) tachycardia has low sensitivity as a diagnostic feature of increasing hypovolaemia in healthy persons. An increase in the pulse rate of 30 beats per minute or more between the supine and standing positions is a highly sensitive and specific sign of hypovolaemia after phlebotomy of large volumes (600–1100 mL) of blood, but the sensitivity is much less after phlebotomy of smaller volumes. The inability to stand long enough for vital signs to be measured because of severe dizziness is a sensitive and specific feature of acute large blood loss. A systolic blood pressure of 95 mmHg or less in the supine position has high specificity but low sensitivity for hypovolaemia. Postural hypotension is present in 10% of normovolaemic people younger than 65 years and in up to 30% of normovolaemic people older than 65 years [8]. The textbook descriptions of the signs of saline depletion in adults (dry mucous membranes, shrivelled tongue, sunken eyes, decreased skin turgor, weakness, confusion) are neither specific nor sensitive compared to laboratory tests for hypovolaemia. The presence of a dry axilla argues somewhat for the presence of saline depletion; the absence of tongue furrows and the presence of moist mucous membranes argue against the presence of saline depletion. The central venous pressure (CVP) is an indicator of the vena caval or right atrial pressure. A vertical distance greater than 3 cm between the top of the jugular venous pulsation (using the external jugular vein or internal jugular vein) and the sternal angle indicates that the CVP is elevated. An elevated venous pressure in persons with pulmonary crackles or peripheral oedema means that the intravascular volume is greater than normal. The absence of visible venous pulsation in the neck veins when the patient is supine or in a head down position indicates significant intravascular volume depletion. The presence of visible venous pulsations in the neck at or below the level of the sternal angle that is seen only when the patient is supine indicates that the intravascular volume is below normal. Acute renal infarction is caused by dissection of the aorta or renal artery, embolism, renal artery thrombosis, renal vein thrombosis or renal artery aneurysm. Acute arterial occlusion is usually symptomatic, with the development of pain (loin, abdominal or back pain), haematuria, proteinuria, nausea and vomiting. Vascular occlusion of a single functioning kidney produces anuria. Atheromatous disease of the renal arteries is common in persons older than 50 years with widespread atherosclerosis. Persons with stenosis or occlusion of one or both renal arteries can develop an elevation in SCr concentration after starting treatment with ACE or ARB drugs or develop acute on chronic renal failure. TMA is a syndrome of microangiopathic haemolytic anaemia, thrombocytopaenia and varying degrees of organ injury caused by platelet thrombosis in the microcirculation. There are two clinically distinct entities: haemolytic uraemic syndrome (HUS) and thrombotic thrombocytopaenic purpura (TTP). HUS affects young children and causes AKI with absent or minimal neurological abnormalities. TTP occurs in adults and causes severe neurological involvement in most cases and variable degrees of renal damage. Both conditions are rare. It can be difficult to distinguish between chronic and acute renal impairment. The following features suggest the presence of chronic renal failure: documented renal impairment in the past, family history of renal disease, polyuria or nocturia, uraemic pigmentation, normochromic and normocytic anaemia or small kidneys on ultrasound or computed tomography (CT) scans. Renal size may be normal or increased in chronic renal failure associated with diabetes, polycystic kidney disease or amyloidosis. The symptoms and signs of urinary tract obstruction depend upon the site, the cause and the rapidity with which it develops. Pain is more common in acute obstruction and is felt in the lower back, flank or suprapubic region, depending on the level of the obstruction. Chronic obstruction is usually painless. Symptoms of prostatic obstruction include frequency, nocturia, hesitancy, post-void dribbling, poor urinary stream and incontinence. Bladder neck obstruction usually results in an enlarged (and palpable) bladder. Muscle necrosis releases intracellular contents into the circulation. This causes red-brown urine (that tests positive for haem in the absence of visible red cells on microscopy or tests positive for myoglobin with specific tests), pigmented granular casts in the urine, elevated serum creatine kinase (CK) levels that are five times or more above the upper limit of normal and clear serum (serum is reddish in haemolysis). The severity of the rhabdomyolysis ranges from asymptomatic elevations of muscle enzymes in the serum to AKI and life-threatening electrolyte imbalances. Urine dipstick findings may be normal because myoglobin is renally cleared from the serum more rapidly than CK, thus myoglobinuria may be absent in patients with renal failure or those who present later in the illness. Muscle pain is absent in about 50% of cases and muscle swelling is an uncommon finding. Muscle weakness occurs in those with severe muscle damage. Fluid sequestration in muscles can cause hypovolaemia. Marked muscle swelling can cause a compartment syndrome. Other blood test abnormalities include hyperkalaemia, AKI with rapid and marked elevation in SCr (e.g. 220 μmol/L per day), hypocalcaemia (which occurs early and is usually asymptomatic), hyperuricaemia, hyperphosphataemia, metabolic acidosis and disseminated intravascular coagulopathy [7]. The early stages of AKI are usually asymptomatic and the diagnosis is based on an elevated SCr concentration. It may take 24 hours or more for an initially normal SCr concentration to show a definite increase and up to 48 hours after the event(s) that caused the AKI to distinguish between the early stages of AKI (risk and injury) and the development of renal failure. The urine output usually decreases and the patient may be oliguric (urine output less than 400 mL per day) or anuric (urine output less than 100 mL per day). Persons with AKI and oliguria have more severe kidney impairment than those without oliguria. Only a few conditions cause complete anuria: total obstruction, vascular lesions, severe ATN or rapidly progressive glomerulonephritis. The clinical features caused by ARF are shown in Table 10.1.4. Table 10.1.4 Clinical features of acute renal failure 1. Anorexia, fatigue, confusion, drowsiness, nausea and vomiting, and pruritus 2. Signs of salt and water retention in the intravascular and interstitial spaces: an elevated jugular venous pressure, peripheral oedema, pulmonary congestion, acute pulmonary oedema 3. Abnormal plasma electrolyte concentrations, particularly hyperkalaemia 4. Metabolic acidosis 5. Anaemia 6. Uraemic syndrome: ileus, asterixis, psychosis, myoclonus, seizures, pericardial disease (pericarditis, pericardial effusion, tamponade) The diagnosis of AKI requires synthesis of data from the patient’s history, physical examination, laboratory studies and urine output. The category of AKI (Risk, Injury or Failure) may be difficult to determine in the emergency department (ED) if the baseline SCr is unknown. The reversibility of the AKI may be inferred if there is a marked increase in urine output after correction of prerenal problems, but a reduction in SCr (due to an increase in GFR) may not be seen for 12–24 hours. The following are measured: serum concentration of electrolytes (sodium, potassium, bicarbonate, chloride, calcium, phosphate), serum urea and SCr concentrations, random blood glucose, liver function tests, coagulation tests and CK concentration. AKI causes acute elevation in the SCr concentration or serum urea concentrations or both. In prerenal AKI, the low urine flow rate favours urea reabsorption out of proportion to decreases in GFR, resulting in a disproportionate rise of serum urea concentration or blood urea nitrogen (BUN) concentration relative to the SCr concentration. However, serum urea concentrations depend on nitrogen balance, liver function and renal function. Severe liver disease and protein malnutrition reduce urea production, resulting in a low serum urea concentration. Increased dietary protein, gastrointestinal haemorrhage, catabolic states (e.g. infection, trauma) and some medications (corticosteroids) increase urea production and increase serum urea concentration without any change in GFR. The SCr concentration is the best available guide to the GFR. Acute reductions in GFR produce an increase in the SCr concentration. The changes in SCr concentration lag behind the change in GFR and can be affected by the dilutional effect of intravenous fluid. Correct interpretation of the SCr concentration extends beyond just knowing the normal values (Fig. 10.1.3). Creatinine is a metabolic product of creatine and phosphocreatine, which are found almost exclusively in skeletal muscle. The SCr concentration is affected by the muscle mass, meat intake, GFR, tubular secretion (which can vary in the same individual and increases as the GFR decreases) and breakdown of creatinine in the bowel (which increases in chronic renal failure). The GFR decreases by 1% per year after 40 years of age, yet the SCr concentration remains unchanged because the decrease in muscle mass with age reduces the production of creatinine. The GFR (corrected for body surface area) is 10% greater in males than females, but men have a higher muscle mass per kilogram of body weight. The SCr concentration in men is thus greater than in women. The creatinine clearance (CCr) or GFR are estimated indirectly using formulae (Cockcroft–Gault formula or the modification of diet in renal disease (MDRD) study equation) based on the SCr concentration (Fig. 10.1.4) [9]. These equations assume a steady-state SCr concentration and are inaccurate if the GFR is changing rapidly. They will also be less accurate in amputees, very small or very large persons or persons with muscle-wasting diseases. Knowledge of a patient’s baseline SCr concentration is important in assessing the severity and progression of AKI. Small changes when the baseline SCr concentration is low are more important than larger changes when the baseline SCr concentration is high. Major decreases in GFR can occur in the normal range of SCr concentration. If the previous SCr concentration is not known, the MDRD equation can estimate the expected (normal) SCr concentration (using a value for the GFR at the lower range of normal). Hyperkalaemia is a common complication, with the serum K+ usually rising by 0.5 mmol/L/day in ARF. The serum Ca2+ concentration may be normal or reduced in ARF. Both hypocalcaemia and hypercalcaemia may occur at different stages of ARF in rhabdomyolysis. Rhabdomyolysis is characterized by a very high blood CK concentration. Abnormal liver function tests invariably accompany the hepatorenal syndrome associated with hepatic cirrhosis. Anaemia develops rapidly in ARF, but its presence or the degree of anaemia does not reliably distinguish between acute and chronic renal failure. Leucocytosis is usually seen if sepsis is the cause of ARF. Eosinophilia is often present in acute interstitial nephritis, polyarteritis nodosa and atheroembolic disease. Anaemia and rouleaux formation suggest a plasma cell dyscrasia. Disseminated intravascular coagulation can complicate ARF due to rhabdomyolysis. A microangiopathic blood film associated with ARF occurs in vasculitis or thrombotic thrombocytopenic purpura. Tests for the detection of antinuclear antibody (ANA) or antineutrophil cytoplasmic antibody (ANCA) or measurement of complement concentration are indicated in suspected cases of vasculitis or glomerulonephritis. The results of urine analysis may be normal in AKI. A positive test for leucocytes, nitrates or both is found in urinary tract infections. A positive test for blood, protein or both suggests a renal inflammatory process. The presence of red cell casts on microscopy is diagnostic of glomerulonephritis. The measurement of the concentration of electrolytes in the urine and the calculation of their fractional excretion is of intellectual interest in understanding the pathophysiological responses of the nephron to different types of AKI. The calculations are cumbersome, the results are inconsistent and the information obtained does not alter the patient’s immediate treatment. A chest X-ray is taken to assess the heart size and the presence of cardiac failure, infection, malignancy or other abnormalities. Ultrasound can define renal size and demonstrate calyceal dilation and hydronephrosis, but the findings depend on the expertise of the operator. Obtaining adequate images is difficult in obese patients, in ascites or where there is a large quantity of gas within the bowel. Ultrasound also provides information about bladder size and can detect prostamegaly. A normal ultrasound examination can occur in the very early stages of obstruction or if ureteric obstruction is due to retroperitoneal fibrosis or to infiltration by tumour. Hydronephrosis not due to obstruction occurs in pregnancy, vesicoureteric reflux or in diabetes insipidus. Doppler scans are useful for detecting the presence and nature of renal blood flow in thromboembolism or renovascular disease; however, because renal blood flow is reduced in prerenal or intrarenal AKI, test findings are of little use in the diagnosis of AKI. CT scans of the urinary tract evaluate renal size and renal position, renal masses, renal calculi, the collecting system and the bladder. Non-contrast CT is the examination of choice in persons with suspected renal calculi and can be used to assess the urinary tract in persons at risk of radiocontrast AKI. Injection of intravenous contrast is used for CT urography, CT angiography and CT venography, which may be necessary in some circumstances. Radionuclide can be used to assess renal blood flow and tubular function. A renal biopsy provides a tissue diagnosis of the intrarenal cause of AKI and is indicated if the findings will identify a treatable condition. A renal biopsy is also valuable when renal function does not recover after several weeks of ARF and a prognosis is required for long-term management. The basis of emergency management is recognizing that AKI is present, correcting reversible factors, providing haemodynamic support and treating life-threatening complications. This is followed by treatment (if available) of the specific cause of AKI and management of ARF by supportive measures and (if required) renal replacement treatment. Hypovolaemia not only causes AKI but also worsens all forms of AKI. The clinical diagnosis of hypovolaemia can be difficult if the jugular venous pressure is not easily seen or if there is pre-existing cardiac failure. When there are definite signs of hypovolaemia, the patient is resuscitated with rapid infusion of crystalloid. If hypovolaemia is a possibility, or if the person’s urine output has decreased markedly, the patient should have 250–500 mL of crystalloid infused rapidly (fluid challenge) and the response (urine output, vital signs, jugular venous pressure) evaluated. An increase in urine output or an increase in blood pressure following a fluid challenge suggests that hypovolaemia was present. Invasive measurement of volume status using central venous and pulmonary artery catheters can increase mortality, lengthen hospital stay and increase the cost of care. There is no evidence to justify the routine use of these invasive measures in patients with AKI. The main indications for central venous cannulation in AKI in the ED are difficulties obtaining intravascular access in the limbs or the need to give drugs which can only be given into a large central vein (e.g. noradrenaline). AKI impairs autoregulation of GFR and renal blood flow throughout all ranges of mean arterial pressure. Renal perfusion in ATN is linearly dependent on mean arterial pressure even in the normal range of blood pressure. Episodes of mild or severe decrease in blood pressure lead to recurrent ischaemic injury. Inotrope/vasopressor drugs (noradrenaline or adrenaline) should be commenced if hypotension persists after correction of hypovolaemia. Dopamine appears to have no clinical advantage compared to other agents and has, in fact, resulted in increased mortality in some studies. Accurate measurement of urine output requires insertion of a urinary catheter, but this is not needed in the less severe forms of AKI if there is frequent spontaneous voiding. A catheter is required initially in persons with oliguria or (apparent) anuria, shock or obstruction to bladder outflow. Frusemide is used to produce a diuresis in the treatment of AKI due to hypercalcaemia and in the treatment of severe rhabdomyolysis. A trial of high-dose frusemide (80–120 mg intravenously) can be used in persons with AKI who have acute pulmonary oedema if dialysis is not readily available. Persons with less severe forms of AKI (e.g. Risk or Injury) who have a low urine output (less than 0.5 mL/kg/h) that does not increase after correction of hypovolaemia are often given low doses of frusemide (e.g. 20–40 mg intravenously). A subsequent increase in urine output is not necessarily associated with a decrease in the SCr concentration. There is no evidence that the use of diuretics to convert the less severe forms of AKI from a (presumed) oliguric to a non-oliguric stage affects outcome [11]. The serum potassium concentration may be low, normal or high. AKI due to diarrhoea causes hypokalaemia and metabolic acidosis, while AKI due to vomiting or diuretics causes hypokalaemia with metabolic alkalosis. A serum [K+] less than 3.0 mmol/L is treated with oral or intravenous potassium. Diabetic ketoacidosis (DKA) causes renal loss of K+, depleting the body of potassium. Persons with AKI due to DKA who have a normal or low serum [K+] need intravenous potassium during treatment with intravenous fluids and insulin. Hyperkalaemia is due to an imbalance between potassium intake and renal potassium excretion or follows redistribution of potassium from the intracellular to the extracellular space. Hyperkalaemia in AKI can be asymptomatic, produce electrocardiogram (ECG) changes or cause potentially fatal changes in cardiac rhythm. The initial ECG changes in hyperkalaemia are shortening of the PR and QT interval, followed by peaked T waves that are most prominent in leads II, III and V2 through V4 (Fig. 10.1.5). Marked ST-T segment elevation (pseudomyocardial infarction pattern) may occur. Bradycardia with sinoatrial (SA) block or atrioventricular block (including complete heart block) can develop and progress to periods of cardiac standstill or asystole. More commonly, the PR interval is prolonged and the QRS complex is widened, with the QRS complex having a left or right bundle branch block configuration (Fig. 10.1.6). At high serum [K+] (8–9 mmol/L), the sinoatrial (SA) node may stimulate the ventricles without ECG evidence of atrial activity (sinoventricular rhythm). When the serum [K+] is 10 mmol/L or greater, SA conduction no longer occurs and junctional rhythms are seen. The QRS complex width continues to increase and, eventually, the QRS complexes and the T wave blend, producing a sine wave ECG. At this stage ventricular fibrillation or asystole are imminent [10].
Genitourinary Emergencies
10.1 Acute kidney injury
Introduction
Stage
Serum creatinine (SCr) concentration
Urine output
RISK
Increase of 1.5 times the baseline
<0.5 mL/kg/h for 6 h
INJURY
Increase of 2.0 times the baseline
<0.5 mL/kg/h for 12 h
FAILURE
Increase of 3.0 times the baseline or SCr is 355 μmol/L or more when there has been an acute rise of greater than 44 μmol/L for 24 h or anuria for 12 h
<0.3 mL/kg/h
LOSS
Persistent acute renal failure; complete loss of kidney function for longer than 4 weeks
END-STAGE RENAL DISEASE
End-stage renal disease for longer than 3 months
Aetiology and pathogenesis
Prerenal acute kidney injury
Renal acute kidney injury
Post-renal acute kidney injury
Epidemiology
Prevention
Maintaining intravascular volume and renal perfusion
Rhabdomyolysis
Radiocontrast nephropathy
Clinical features
Evaluation of prerenal (intravascular volume) status
Evaluation of the renovascular state
Exclusion of thrombotic microangiopathy (TMA)
Pre-existing renal disease or chronic renal failure
Exclusion of urinary obstruction
Recognition of rhabdomyolysis
Acute kidney injury and acute renal failure
Differential diagnosis
Criteria for diagnosis
Serum biochemistry
Full blood examination
Serological tests
Urine tests
Imaging
Renal biopsy
Treatment
Correction of hypovolaemia
Haemodynamic support
Monitoring and maintaining urine output
Urinary Catheter
Diuretics
Electrolyte abnormalities
Potassium
< div class='tao-gold-member'>
Related posts:

Full access? Get Clinical Tree


10. Genitourinary Emergencies

Only gold members can continue reading. Log In or Register to continue
